• MC-F-008. Capítulo 8. Daños en el genoma y el envejecimiento.
8.1 Introducción
Las teorías que consideran el daño molecular como origen de procesos degenerativos asociados al envejecimiento (Harman, 1991) parecen explicar, en gran medida, muchos de los fenómenos relacionados con este. Según dichas teorías cabe esperar que el deterioro en las estructuras moleculares aumente con el tiempo, a pesar de que las células cuentan con dispositivos muy eficientes para contrarrestarlos a medida que son detectados. Las manifestaciones fenotípicas de esas alteraciones serán más o menos acentuadas en función de la cantidad y el tipo de moléculas afectadas. En este sentido hay que pensar que los daños en biomoléculas (proteínas, lípidos, etc.) tendrían, en principio, escasa repercusión si se producen a baja frecuencia, ya que según el tipo de molécula dañada podrían ser reparada o sintetizada una nueva copia de la misma. No ocurriría lo mismo si el daño se produjese en determinadas regiones del genoma de células somáticas o germinales (se incluyen en esta denominación genérica tanto células de tejidos reproductivos como las células madre de tejidos adultos) que contengan genes o regiones reguladoras de la expresión de los mismos, ya que en este caso, si el daño permanece sin reparar podría verse afectada fenotípicamente la línea celular en cuyo genoma se localice la alteración (Lombard et al., 2005).
Dada la relativa facilidad con la que hoy se puede amplificar y analizar una determinada región del genoma, no resulta extraño que en los últimos años se hayan incrementado de manera considerable los trabajos encaminados a la búsqueda de genes implicados en la etiología molecular de numerosas enfermedades, y entre estas las que se asocian al envejecimiento. En este sentido los casos más representativos tal vez sean los tumores y determinados procesos degenerativos, debido al incremento de la incidencia de los mismos en relación con la edad y su etiología mutacional. La relación causal entre deterioro genómico y envejecimiento parece evidente (Johnson et al, 1999), y tal vez sea esta la faceta mejor estudiada a nivel molecular en lo que al envejecimiento se refiere, por ello dedicamos este capítulo a exponer las causas del deterioro del ADN celular y los mecanismos de que dispone la célula para repararlo.
8.1.1 Inestabilidad del material genético
Cuando a lo largo de la evolución la naturaleza selecciona una determinada molécula para desempeñar una función concreta, se supone que se trata del instrumento óptimo para ello, aunque en algunos casos resulta difícil conjugar ciertas propiedades como eficiencia funcional y estabilidad química, un ejemplo claro de esta dificultad la tenemos en el caso de los ácidos nucleicos como portadores del mensaje genético. Las bases nitrogenadas que forman parte del ADN son especies moleculares relativamente inestables, y en el medio intracelular sufren, de manera espontanea y con relativa frecuencia, modificaciones químicas (Figura 1 y Tabla 1) (Watson et al., 1987). La composición química de los nucleótidos también se puede ver modificada por la acción de agentes externos (mutágenos) de naturaleza física o química, aunque este tipo de alteraciones no serán tratadas en este trabajo, hay que decir que muchas de ellas son detectadas y reparadas por los mismos mecanismos celulares utilizados para el caso de las alteraciones espontáneas.
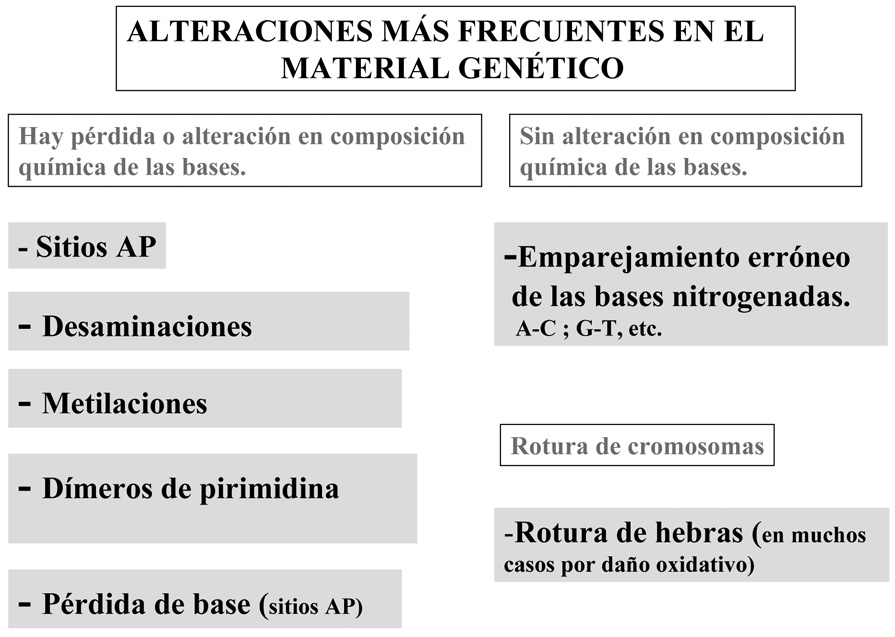
Figura 1. Modificaciones más frecuentes en la copia del ADN.
Otro posible origen de alteraciones espontaneas en el ADN radica en la gran similitud estructural que presentan las diferentes bases entre sí (púricas o pirimidínicas), esto hace que en algunas ocasiones la maquinaria encargada de duplicar el material genético, a pesar de su elevada eficiencia en el proceso, “confunda” las bases y origine una mutación. Aún así la precisión con que la maquinaria replicativa lleva a cabo su función es muy alta, ya que además dispone de un mecanismo de supervisión y corrección de la copia recién sintetizada. Se calcula que la frecuencia con la que la maquinaria replicativa coloca en el ADN sintetizado una base incorrecta es de aproximadamente una de cada 10.000, cifra importante si tenemos en cuenta el número total de nucleótidos que tiene un cromosoma eucariótico. No obstante el sistema de revisión de copia de la polimerasa rebaja la frecuencia de errores unas 100.000 veces sobre la anterior, por lo que podemos decir que la frecuencia de errores espontáneos cometidos por la polimerasa nuclear encargada de replicar el ADN es de aproximadamente un nucleótido de cada 109. Este no sería el caso de la polimerasa del ADN mitocondrial, donde la eficiencia a la hora de corregir errores es mucho más baja, lo que se traduce en una tasa mutacional más elevada que la que se observa en el ADN nuclear.
De lo anteriormente expuesto se concluye que, según la fase del ciclo celular que consideremos, hay dos tipos de mutaciones en función de su origen:
- A) Las que se producen sobre el ADN no replicante producto de alteraciones químicas (espontáneas o inducidas por agentes internos o externos) en las bases nitrogenadas.
- B) Las que se producen en la fase S, originadas por un error de la maquinaria replicativa.
Las mutaciones que más parecen afectar a la célula son aquellas que, no habiendo sido previamente reparadas, obstruyen el proceso replicativo o la transcripción genética, por ello los mecanismos de reparación más activos los encontraremos actuando en las etapas del ciclo celular donde ocurran dichos fenómenos. Se supone que el ADN presente en la cromatina de alto grado de compactación (heterocromatina) no sería objeto de supervisión frecuente fuera de la fase S del ciclo por dos razones:
- A) El recubrimiento proteico del ADN en la cromatina disminuiría la posibilidad de que aquél se exponga a agentes mutagénicos, reduciéndose de esta forma la frecuencia de mutación (en este caso el ADN mitocondrial, al carecer prácticamente de histonas, estaría más expuesto a la acción de agentes mutagénicos).
- B) La alta compactación de la cromatina impediría a la maquinaria de supervisión de la copia acceder a la superficie del ADN para poder realizar su labor. No obstante es mucho lo que queda por conocer a este respecto.
Aunque la mutabilidad de la copia del material genético podría considerarse una circunstancia adversa para la célula y su descendencia, es esta “naturaleza mutable” del material genético lo que ha permitido evolucionar a los seres vivos por adaptación progresiva a las condiciones impuestas por un entorno variable. En algunos casos la forma en la que se manifiesta una mutación es una alteración morfológica o funcional. En otros casos su nivel de letalidad es tal que nunca dará lugar a un sujeto viable. Aunque, en la mayoría de los casos, las mutaciones puntuales no dejan una huella fenotípica (mutaciones silenciosas), bien sea porque no tienen como consecuencia la modificación de la secuencia de aminoácidos de la proteína codificada por el gen mutado, o porque la sustitución de un aminoácido por otro no altera mucho la función de dicho producto. En el ámbito de estudio en el que nos encontramos –la senescencia– tenemos que hacer además una clara distinción entre aquellas mutaciones que heredamos de los progenitores y las que adquirimos a lo largo de la vida, y que mayoritariamente afectan a nuestras células somáticas. Entre las primeras podría haber algunas que nos predispusiesen al envejecimiento prematuro (disfunción acentuada de un tejido específico aún en fases tempranas de la vida), y que darían lugar a una serie de patologías que se conocen con el nombre genérico de “progerias” (precisamente en muchas de ellas están mutados genes que defienden al organismo frente a las mutaciones). Las segundas se irían adquiriendo de manera aleatoria a lo largo de la vida, en función de la presión del ambiente, y su acumulación iría reduciendo progresivamente la funcionalidad de los distintos órganos y tejidos hasta alterar la homeostasis orgánica, de tal forma que a partir de cierto grado de deterioro haría inviable el sistema produciéndose como consecuencia la muerte. Como vemos en esta última definición va implícita la de envejecimiento.
8.2 Modificaciones más frecuentes en la copia del ADN
8.2.1 Sitios “AP”
Genéricamente se entiende por “sitio AP” un punto del genoma (ADN) que ha sufrido la pérdida espontánea de la base nitrogenada; en este caso la base se “desengancha” de la ribosa generándose un sitio (AP) “apurínico” o “apirimidínico” (según la base que se pierda). Se calcula que en una célula humana se generan al día unos 5.000 sitios AP. La polimerasa encuentra dificultades para replicar una zona donde hay un sitio AP, por lo que la alteración debería repararse antes de que la horquilla replicativa o la maquinaria transcripcional alcance esa zona.
8.2.2 Desaminaciones
Otro tipo de alteraciones que sufre el ADN es la pérdida de grupos amino de sus bases nitrogenadas (las que lo tienen: A, G o C) (Figura 2). Hay que tener en cuenta que la desaminación de una citosina produce uracilo, esta base no existe en el ADN pero sí en el ARN, y por ello ha de ser sustituida nuevamente por una citosina.
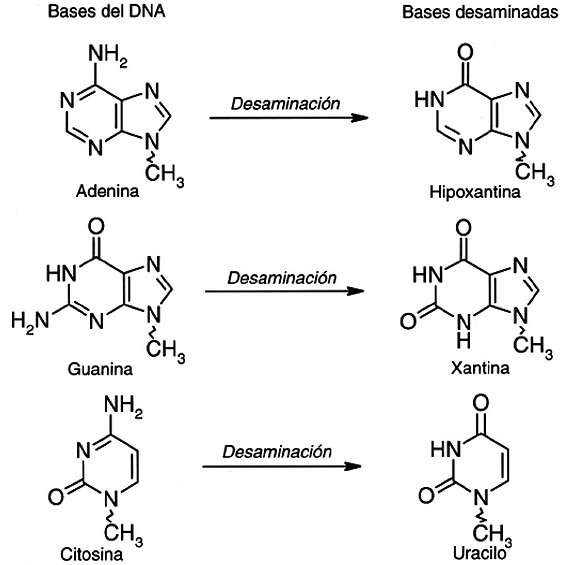
Figura 2. Cambio de composición de bases nitrogenadas por desaminación.
Un caso relevante en las desaminaciones de las bases surge como consecuencia de la desaminación de la 5-metil-citosina, en este caso la retirada del grupo amino de la 5-metil-citosina convierte a esta en timina, que es un nucleótido normal en el ADN, por lo que si este error no se repara antes de la replicación la maquinaria encargada de la misma podría formar una hebra hija con una A en lugar de una G. La importancia de este tipo de desaminaciones se debe a la frecuencia con la que aparecen en el genoma eucariótico pares GC en los que la citosina está metilada. Se piensa que la frecuencia de desaminaciones por célula y día es más baja que la de formación de sitios AP, pero no por ello dejan de ser una causa importante de mutaciones.
Tanto los sitios AP como las desaminaciones son consecuencia directa de la naturaleza inestable del nucleótido, estas modificaciones están desencadenadas por variaciones en el entorno fisico-químico de la molécula que suponen cambios de pH o de temperatura, cuando estos se apartan de los valores fisiológicos normales.
8.2.3 Metilaciones
En algunos casos se producen en el ADN metilaciones, que consisten en la introducción de grupos metilo (–CH3) en un átomo de la base nitrogenada. La metilación de bases nitrogenadas del ADN es un fenómeno habitual tanto en procariotas como en eucariotas, ya que dicha modificación específica (como es el caso de la metilación de restos de citosina en los pares CG en eucariotas) cumple una función en la célula. No obstante se conocen metilaciones atípicas de las bases, como en el caso de la formación de un derivado metilado de la guanina (O6-metil-guanina). Esta base modificada puede generar una mutación post-replicativa si no es reparada a tiempo, ya que aparea con la misma probabilidad C y T.
|
INESTABILIDAD QUÍMICA PROPIA DEL ADN Pérdida de base, metilaciones, desaminaciones, etc. |
|
SUBPRODUCTOS DEL METABOLISMO CELULAR Radicales libres y otros oxidantes. |
|
AGENTES EXTERNOS Radiaciones U.V., radiaciones ionizantes, agentes genotóxicos. |
|
ERRORES DE LA MÁQUINA REPLICATIVA Colocación defectuosa de bases en la replicación del ADN. |
Tabla 1. Origen de las alteraciones en el ADN.
8.2.4 Dímeros de pirimidina
Este tipo de alteración es frecuente encontrarla en células de la epidermis, y se caracteriza por la formación de un enlace covalente entre dos bases pirimidínicas colocadas en posiciones adyacentes en el ADN. Suelen formarse cuando el ADN se expone a una fuente de radiación U.V. (Figura 3).
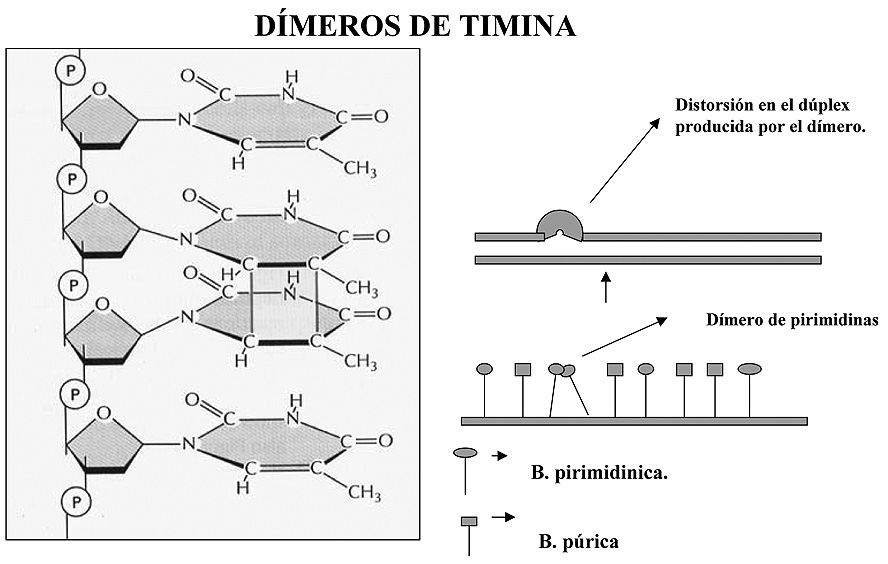
Figura 3. Formación de dímeros de timina. La formación de un enlace entre dos restos de timina adyacente distorsiona la estructura de la doble hebra.
La formación de un enlace entre ambas bases distorsiona la doble hebra de tal forma que compromete la replicación o la transcripción, por lo que han de ser reparados antes de que estas concluyan. El hecho de que solamente se vea afectado tejido epidérmico se debe a que la capacidad de penetración de la radiación U.V. no va más allá de unos milímetros.
8.2.5 Daño oxidativo en el ADN y rotura de hebras
Otro tipo de daños, no poco frecuentes en el material genético, son las reacciones químicas que se producen entre componentes del ADN y agentes químicos muy activos producidos por la propia actividad celular (Halliwell y Gutteridge, 1999; Hasty y Vijg, 2002). En este sentido las especies reactivas del oxigeno (RLO) –que hemos descrito en el capítulo anterior– serían los ejemplos más representativos de dichas sustancias. La interacción de los OH. con el ADN deja toda una serie de “huellas químicas” donde la más caraterística es la 8-oxo-2’ deoxiguanina (Zglinicki et al., 2001), por lo que resulta relativamente fácil cuantificar el daño evaluando la presencia de ese compuesto. Además de esto, los radicales OH. actúan como fuertes oxidantes capaces de reaccionar no sólo con las bases nitrogenadas, sino que pueden hacerlo, aunque en menor grado, con otros elementos moleculares del dúplex, pudiendo provocar con ello la rotura de una o ambas hebras del ADN (Barnes, 2002), lo que supone un daño considerable en el genoma. Se estima que el número de ataques de especies reactivas del oxígeno sobre el ADN por célula y día es de aproximadamente 100.000 en el ratón y de unos 10.000 en humanos. Llegado este punto hay que hacer una matización de interés, y esta se refiere al hecho de que la única especie reactiva del oxígeno que parece que es capaz de atacar diferentes componentes moleculares del ADN (azúcar o base nitrogenada) es el radical OH.. Como ya se comentó anteriormente, la presencia de este radical en el entorno del ADN en elevadas concentraciones, puede provocar la rotura de la doble hebra. Esto es lo que ocurre en el caso de que radiaciones ionizantes incidan sobre el ADN, ya que estas generan radical hidroxilo a partir del agua presente en el organismo. Se descarta, al menos en condiciones normales, que tanto el radical O2.- como H2O2 y NO. dañen directamente al ADN, sino que lo harían previa generación de radical hidroxilo (muy probablemente por reacción de Fenton en el caso del H2O2). Teniendo en cuenta que la mayoría de derivados oxidantes del oxígeno se producen en la mitocondria, y dada la alta susceptibilidad de su ADN a la mutación por diferentes razones (falta de histonas, falta de mecanismos reparadores eficientes, etc.), no es extraño que la tasa de mutaciones en el genoma mitocondrial sea considerable.
Los experimentos in vitro realizados con células eucarióticas tratadas con H2O2 han demostrado que las mutaciones producidas por este agente químico se tratan de deleciones y sustituciones de bases. Además, la distribución de estas mutaciones no parece que sea aleatoria, ya que hay zonas del genoma que parecen ser más susceptibles que otras. En principio no hay una explicación razonable a este comportamiento, pero podría estar relacionado con el grado de compactación de la cromatina en la zona mutada o la interacción de dichas regiones con metales de transición implicados en la reacción de Fenton y que generarían radicales OH. a partir de H2O2.
8.3 Mecanismos de reparación del ADN
Como acabamos de ver, la estabilidad de la copia del material genético en un entorno celular considerado como normal está muy comprometida, por lo que si no hay nada que se oponga a ello, el número de mutaciones por célula y generación sería de tal calibre que la vida resultaría difícil o imposible en tales circunstancias. Sin embargo, como ya se adelantó, la célula ha adquirido a lo largo de la evolución toda una serie de mecanismos que tienden a reducir el daño que se produce en el ADN (Wood, 1996). En ocasiones estos actúan haciendo desaparecer el agente inductor del daño, como es el caso de las enzimas antioxidantes: superóxido dismutasa (SOD), catalasa, etc., neutralizando de esta forma especies reactivas del oxígeno, que de otra forma podrían atacar el ADN, mientras que otros mecanismos (tal vez los más variados) tratarían de reparar el daño producido en esta molécula. Las características generales de estos sistemas de reparación aparecen comentadas en la Tabla 2.
|
– Ubicuidad: en todas las células. – Redundancia: varios sistemas por célula. – Complejidad: numerosos elementos intercambiables en la mayoría de los casos. – Eficiencia: nunca al 100% (unos más que otros). – Variabilidad funcional: un mismo sistema podría actuar de manera diferente en cada tipo de célula. – Homología interespecífica: por lo general bien conservados evolutivamente. |
Tabla 2. Características generales de los sistemas de reparación y supervisión del ADN.
La importancia que tiene el mantener una copia “sana” del material genético para la célula, ha hecho que desde una etapa muy temprana, los seres vivos desarrollasen toda una serie de estrategias para reparar los daños que se producen de manera constante en el ADN. Los primeros estudios que analizaron estos mecanismos se llevaron a cabo en procariotas, más tarde el campo se amplió a eucariotas. En ambos casos se observó que dichos mecanismos son redundantes, es decir que la célula no confía la supervisión y reparación de su genoma a uno sólo de ellos, sino que dispone de varios que en muchos casos interaccionan. Se calcula que una célula de mamífero dispone de al menos 130 genes involucrados en tales funciones, y no resultaría extraño que, a medida que se conozca mejor el papel de los distintos genes humanos, aparezcan nuevos loci relacionados con la reparación del ADN. Estos datos, por sí solos, dan una idea de la importancia que para una célula tiene el mantener su genoma sin defectos. Pero el aspecto más representativo de la importancia de los mecanismos de reparación del ADN tal vez sea el gran número de patologías cuya etiología se asocia a fallos en elementos moleculares en dichos mecanismos.
En la mayoría de los casos, en estos mecanismos intervienen numerosos componentes, por lo que su modo de acción no deja de ser complejo. En nuestro caso presentaremos los más relevantes (Tabla 3), y dentro de cada uno se presenta de forma simplificada su mecanismo de acción.
|
REPARACIÓN DIRECTA Por estos mecanismos se reparan: metilación de guanina, y en algunos vertebrados dímeros de pirimidina. No intervienen nucleasas ni ADN-polimerasas. |
|
REPARACIÓN INDIRECTA Hay intervención de nucleasas y ADN-polimerasas. Se necesita hebra “molde” perteneciente al mismo cromosoma o al homólogo.
|
Tabla 3. Diferentes sistemas de reparación del ADN en eucariotas según su mecanismo de acción.
La etapa más importante del proceso tal vez sea aquella en la que se detecta el punto o puntos del genoma donde se localiza la alteración. Se cree que una de las condiciones para que esto se pueda llevar a cabo es que la molécula de ADN se encuentre descompactada, lo que permitiría al sistema de detección “rastrear” dicha región en busca de errores, que se localizarían por la distorsión estructural que un apareamiento de bases incorrecto produce en el dúplex. En este sentido hay evidencias de que tanto la replicación como la transcripción del ADN son etapas donde se lleva a cabo una intensa actividad por parte de los sistemas de reparación.
Los sistemas de reparación indirecta intervienen sobre el ADN, en replicación (fase S), transcripción o sobre hebras de ADN seccionadas. Como ya se comentó anteriormente, la propia polimerasa del ADN, o algunos de los componentes del mecanismo transcripcional, llevan a cabo la supervisión de la copia recién sintetizada. En otros casos hay complejos moleculares que cooperan con la polimerasa del ADN resolviendo casos en los que esta ha de replicar o transcribir una determinada zona del genoma en la que se detecta una alteración. A pesar de la gran cantidad de mecanismos y moléculas que intervienen en estos procesos hay un denominador común en todos ellos, y es que la mayoría están conservados evolutivamente.
Otros de los mecanismos que actuarían para preservar la integridad del material genético serían aquellos que, en lugar de intervenir reparando la copia del ADN, lo harían supervisando la integridad de los desoxirribonucleótidos que serán utilizados para la síntesis del ADN, ya que algunos derivados de estos pueden ser incorporados por la polimerasa en lugar del d-ribonucleótido normal pudiendo dar lugar a una mutación , tal es el caso del 8-hidroxi-dGTP.
8.3.1 Mecanismos de reparación directa
8.3.1.1 Recuperación de guaninas metiladas
Una de las alteraciones que se conocen en el ADN es el caso de la metilación de restos de guanina para formar O6-metilguanina; en este caso la célula dispone de una enzima “suicida” que localiza el lugar de la alteración y seguidamente transfiere el grupo metilo desde la guanina a un resto de cisteína en su centro activo, la enzima queda ahora inactivada, ya que este proceso no es reversible (Figura 4).
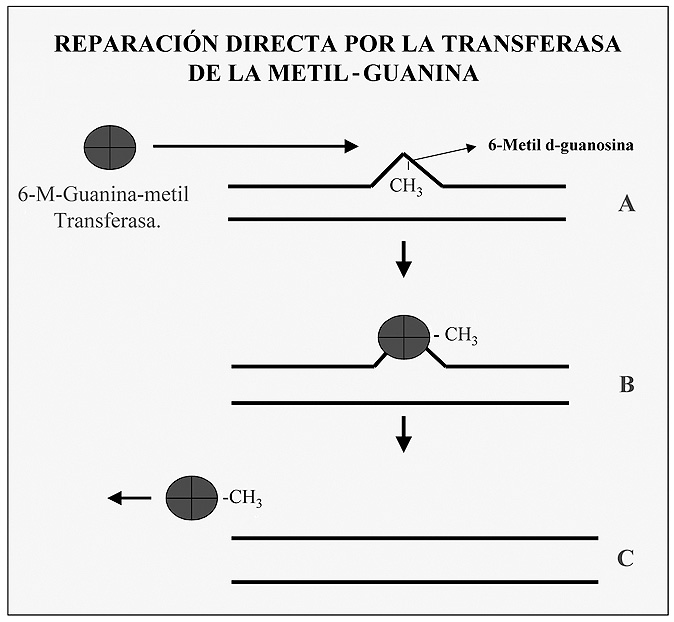
Figura 4. Recuperación de guaninas metiladas por acción de la guanina metil-transferasa.
8.3.1.2 Reparación de dímeros de pirimidina
Un sistema similar actúa en procariotas y en muchos eucariotas, en él interviene una enzima que detecta dímeros de pirimidina que se producen en restos adyacentes C-C, C-T (siendo el más frecuente T-T). En procariotas la enzima que repara esta alteración es la fotoliasa. En primer lugar la enzima localiza el punto donde se ubica el dímero y seguidamente se posiciona sobre él y repara la alteración (Figura 5).
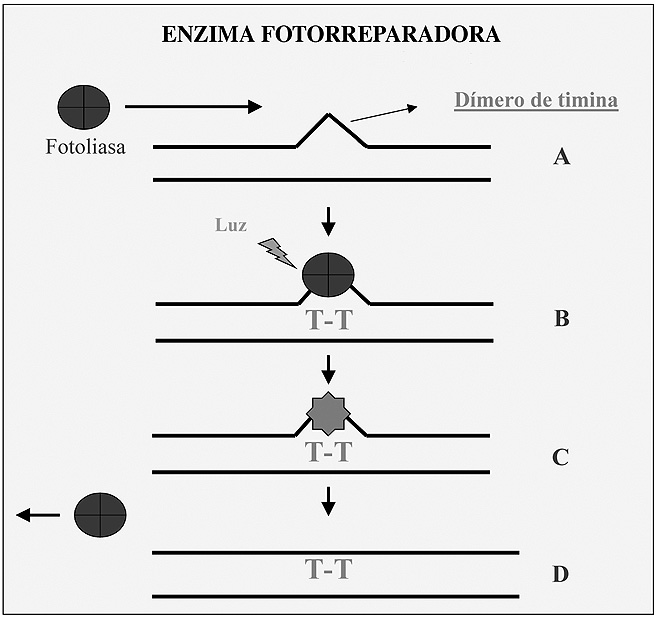
Figura 5. Reparación de dímeros de pirimidina.
Esta enzima además tiene una interesante peculiaridad, y es que se activa por irradiación con longitudes de onda visibles próximas al U.V. Aunque en algunos vertebrados se han detectado enzimas que actúan de manera similar a la fotoliasa no se ha descrito todavía para el caso del ser humano, donde este tipo de alteraciones se repara por otros mecanismos. En este caso, los dímeros de pirimidina (que se supone un tipo de lesión frecuente que produciría el envejecimiento de la piel) se pueden reparar por otro mecanismo (escisión de nucleótido) que se describirá más adelante.
8.3.2 Mecanismos de reparación indirecta
En general estos mecanismos actúan eliminando la base alterada utilizando diferentes estrategias:
8.3.2.1 Reparación por escisión de base
En este caso la base alterada es retirada del ADN por un tipo de enzimas denominados glicosidasas. Hay varias y cada una se ocupa de un tipo de modificación (Hipoxantina-ADN glicosidasa, uracil-ADN -glicosidasa, etc.). Cuando estas retiran la base dañada se genera un sitio AP (también se pueden generar sitios AP por otras causas), que resultaría muy mutagénico si se dejase sin reparar, ya que bloquearía la replicación o transcripción del ADN en ese punto, por lo que seguidamente interviene una endonucleasa que retira el resto de ribosa fosfato del sitio AP, dejando un hueco de un nucleótido que es rellenado inmediatamente por la acción de una polimerasa del ADN, por último la hebra es sellada por la ligasa (Figura 6).
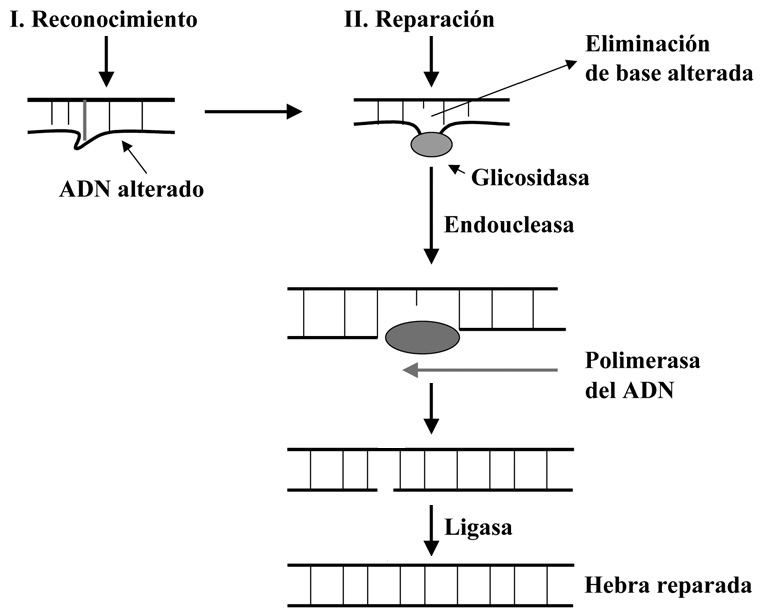
Figura 6. Mecanismo de reparación por escisión de base.
8.3.2.2 Reparación por escisión de nucleótido o hebra
Este mecanismo de reparación es muy similar al anterior, y de hecho comparte con aquél algunos de sus elementos moleculares. En una primera etapa, el sistema reconoce el punto de la lesión. Seguidamente actúa una endonucleasa que corta un pequeño fragmento de la hebra que presenta la lesión a ambos lados de la misma dejando entre el nucleótido afectado y ambos puntos de corte varios nucleótidos. Luego se retira esta porción de la hebra al tiempo que una polimerasa de ADN comienza la síntesis del fragmento que sustituirá al eliminado, tomando como molde la hebra “sana” (Figura 7).
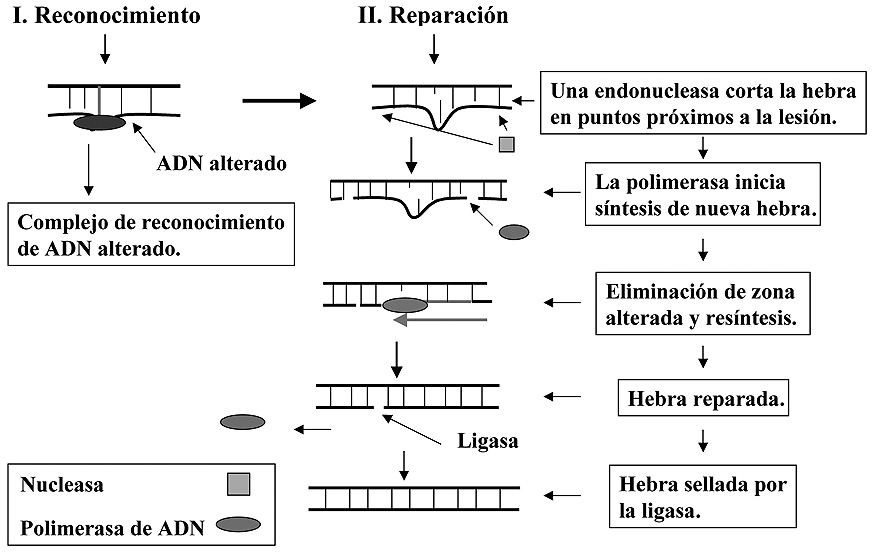
Figura 7. Mecanismo de reparación por escisión de nucleótido.
Como en el caso anterior la ligasa sellará la hebra nueva, dejando de esta forma reparada la alteración (en la descripción de estos mecanismos, y para su simplificación, se omiten otras proteínas que intervienen en el proceso, y que son también importantes para que este se lleve a cabo). Son muchas las alteraciones que se reparan por este mecanismo, entre ellas los dímeros de pirimidina, bases alteradas, etc. En bacterias este sistema está muy bien estudiado y se parece mucho al que actúa en levaduras (eucariotas unicelulares), aunque en estas últimas consta de más elementos y es por ello más complejo. En mamíferos el sistema es similar al de levaduras lo que demuestra que está evolutivamente bien conservado.
8.3.3 Mecanismos de reparación por recombinación
Este mecanismo es complejo, y a diferencia de los hasta ahora estudiados, utiliza una estrategia de recombinación similar a la que opera en el intercambio de cromátidas que se da en la meiosis, uniendo regiones homólogas de los cromosomas paternos y maternos. El tipo de alteraciones que se reparan mediante este mecanismo suelen ser aquellas que, por diferentes razones, no permiten disponer de hebra molde (rotura de hebras, apareamientos anómalos entre bases, etc.). Este último tipo de anomalía ha de ser reparada antes de que la zona sea replicada por la polimerasa, ya que de no ser así se podría bloquear el proceso replicativo en dicho punto o inducir una mutación permanente en la descendencia celular. Si durante la fase S del ciclo celular, la polimerasa de ADN se encuentra una alteración que no ha sido reparada anteriormente, puede ocurrir que introduzca un nucleótido al azar con el consiguiente riesgo de mutación, o continúe su labor dejando sin replicar la zona alterada hasta su reparación. En este último caso la solución al problema consiste en retirar la porción de una de las hebras no replicadas y seguidamente sintetizar una hebra nueva de ADN por un mecanismo que toma como molde la hebra complementaria del cromosoma homólogo (Figura 8). Se han descrito varias polimerasas del ADN que participan en la resolución de problemas de este tipo. En general estas polimerasas no poseen la alta fidelidad de copia de la polimerasa normal (la que replica el genoma en la fase S del ciclo celular), por lo que cabe esperar una tasa de error incrementada con respecto al que presenta esta última, y parece que cada una de ellas se especializa en solucionar un tipo específico de alteración.
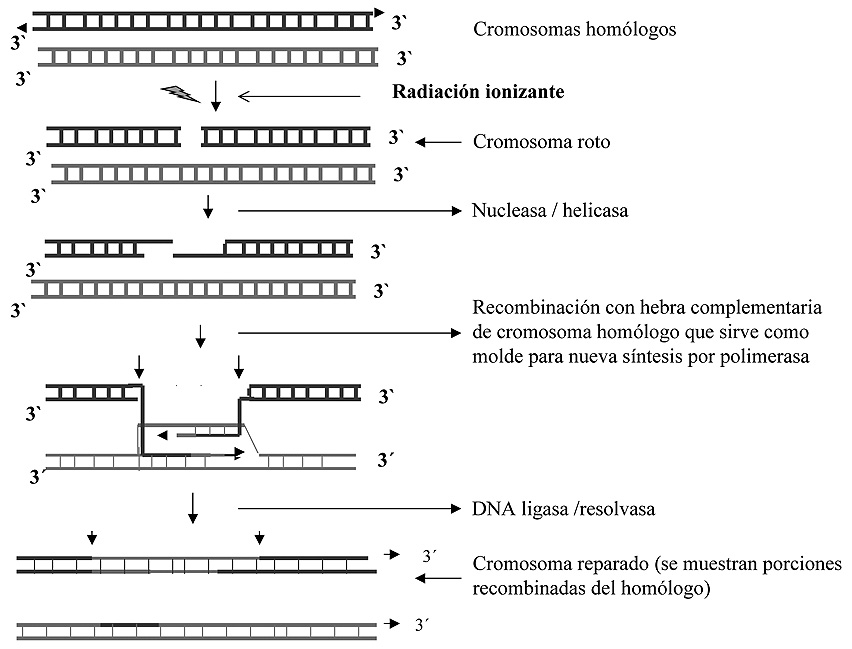
Figura 8. Mecanismo de reparación de cromosomas fragmentados. Reparación mediante recombinación.
En general la fase S del ciclo celular resulta crítica para la vida de la célula, ya que en ella el material genético a replicar ha de estar en las mejores condiciones posibles antes de ser transferida a la descendencia, de tal forma que si dicha copia está muy dañada podría bloquear el proceso replicativo, hasta el punto de que la célula opte por el “suicidio” (apoptosis) para evitar que pasen genes defectuosos a la descendencia. El mismo sistema que regula la entrada en apoptosis alerta a los mecanismos de reparación de errores en la copia del ADN celular a transcribir. En caso de que este sistema falle podrían acumularse un número excesivo de mutaciones en la célula. En este sistema de control participan numerosas proteínas, aunque una de ellas, la denominada p53 juega un papel decisivo en el mismo. Una prueba evidente de la importancia de esta proteína en el control del ciclo celular la tenemos en el hecho de que una de las mutaciones más frecuentes en tumores afecta a dicha proteína, este aspecto se tratará más en detalle en el siguiente apartado.
El sistema de reparación por recombinación homóloga se cree que es utilizado con frecuencia por la célula para llevar a cabo la reparación de cromosomas rotos. Este tipo de lesión, como ya se ha comentado, puede producirse por ataque de radicales hidroxilo sobre el dúplex de ADN. De no repararse, la presencia de tales alteraciones provocará graves daños a la célula, y su descendencia será con toda probabilidad inviable debido a la pérdida de una cantidad importante de material genético. Por ello la célula es particularmente sensible a este tipo de lesión, y para remediarlo pone en marcha complejos mecanismos de reparación en los que intervienen un elevado número de proteínas.
Además de esta estrategia de reparación que se basa en la recombinación homóloga, se sabe que la célula dispone de otra no menos sofisticada (aunque menos estudiada) para reparar el mismo tipo de lesión. En este mecanismo la reparación de la rotura se produce por empalme entre los extremos de los fragmentos del dúplex truncado (Barnes, 2001).
8.4 Reparación de ADN y enfermedad
Tras los recientes hallazgos sobre las bases genéticas y moleculares de muchos procesos degenerativos, se puede decir que las deficiencias en los sistemas de mantenimiento de la integridad del genoma pueden acarrear desórdenes genéticos debilitantes, envejecimiento prematuro, predisposición al cáncer y procesos degenerativos (Shiloh y Lehmann, 2004).
Con toda probabilidad la etapa más importante en el proceso de reparación de los daños inducidos al ADN es la del reconocimiento de la lesión (Kolodner, 2000). En general las lesiones que se reparan con mayor rapidez son aquellas que se localizan en regiones donde el ADN está menos compactado, “más abierto”, esto es, donde se replica o donde ha de transcribirse (hay factores de transcripción que intervienen en mecanismos de reparación del ADN “abierto a transcripción”). Esto tiene su sentido pues ambas funciones son imprescindibles para la vida de la célula y requieren de una copia de ADN en buen estado (Figura 9). Por otra parte las lesiones localizadas en zonas silenciadas (heterocromatina) del genoma no tendrían en principio repercusiones fenotípicas al no expresarse los genes contenidos en ellas.
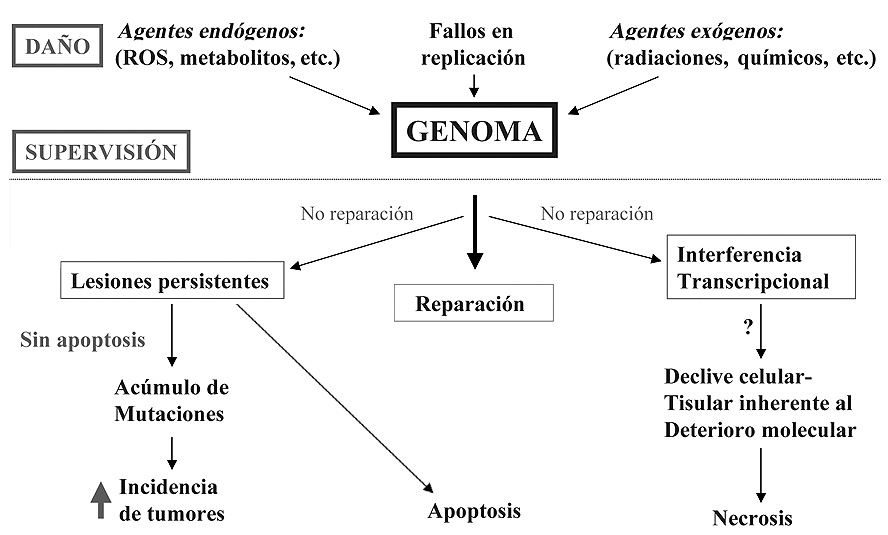
Figura 9. Daño en el genoma y envejecimiento.
Como ya se comentó en el apartado anterior, cuando una célula no puede replicar o transcribir un ADN por el grado de deterioro de la copia, se para temporalmente el ciclo celular, con el fin de dar tiempo a la maquinaria de reparación a que desarrolle su tarea. Cuando la situación es crítica –debida al grado de deterioro del ADN– la célula entra en apoptosis, abortando con ello el ciclo celular, y retirando del sistema la línea celular cuyo genoma se encuentra dañado. Hay proteínas que detectan alteraciones del dúplex o roturas cromosómicas en el tránsito G1–S y activan un complejo mecanismo para repararlas, tal es el caso de las proteínas BRCA, que de alguna forma detectan la presencia de errores en el ADN (interviene en reparaciones de rotura de hebras; las mutaciones en los genes que codifican para estas proteínas predisponen al cáncer de mama y otros tumores) y activa la expresión de p53. Esta proteína (p53) es un factor de transcripción que pone en marcha un mecanismo complejo, que tiene como finalidad el retener el ciclo celular en G1-S mientras se reparan los errores. La proteína p53 regula además la entrada de la célula en apoptosis en el caso de que los daños inducidos al ADN no sean reparados. Se sabe que una proteína denominada ATM (la proteína alterada en la ataxia telangiectasia) se activa cuando el ADN está dañado, y se cree que interviene en la activación de p53.
No obstante los niveles celulares de p53 han de estar bajo estricto control, ya que la hiperexpresión de p53 puede inducir senescencia por retención del ciclo celular o apoptosis, mientras que su carencia, o un defecto en su funcionamiento, permite el paso de numerosas alteraciones genéticas a la descendencia, en estas condiciones aumenta considerablemente el riesgo de transformación tumoral. La relación causa-efecto entre la presencia de genotóxicos y p53 queda patente por el hecho de que los niveles intracelulares de p53 aumentan después de someter a la célula a radiaciones ionizantes o U.V., lo que hace pensar que el estímulo para su síntesis es el incremento del daño al ADN inducido por los RLO que se producen al incidir las radiaciones en el medio celular.
En el caso de que el sistema que regula la entrada en apoptosis no funcione, la célula continúa su ciclo celular independientemente de las lesiones que lleve el genoma, en este caso lo más probable es que esta línea celular acumule con el tiempo una combinación de mutaciones que la conducirán a una incapacidad metabólica o a una transformación maligna, de ahí que la patología que más frecuentemente se asocie al envejecimiento sea el cáncer (Tabla 4) (Ames,1993; Hoeijmakers, 2001; Klaunig y Kamendulis, 2003).
|
– Xeroderma Pigmentosum. – Cáncer. – Ataxia telangiectasia. – Síndrome de Bloom. – Síndrome de Cockaine. – Síndrome de Werner. |
Tabla 4. Enfermedades hereditarias y defectos en la reparación del ADN.
La manifestación más drástica de la importancia que tienen los sistemas de reparación son las patologías asociadas a defectos en locus cuyos productos están involucrados en dichos sistemas, algunos ejemplos serían: el Xeroderma pigmentoso (Wood, 1999) (donde hay, al menos, siete loci diferentes cuya mutación independiente produce la enfermedad), ciertos tipos de cáncer, síndrome de Bloom, síndrome de Werner, síndrome de Cokcayne, tricotiodistrofia, etc.
Sin lugar a dudas el cáncer es una enfermedad de nuestros genes, aunque bajo esta denominación genérica se agrupan diferentes etiologías moleculares. El aumento de la frecuencia de aparición de cáncer con la edad se asocia al aumento de daños en el material genético también con la edad. Estos daños, además de a otros loci, afectarían fundamentalmente a oncogenes y genes supresores de tumores, activando los primeros y silenciando los segundos, en muchos de estos casos hay que decir que no existe mutación en el sentido estricto del concepto (no hay alteración de la secuencia de bases en el ADN), sino más bien un fenómeno epigenético.
Lo que llama la atención en los sistemas de reparación de daños en el ADN es que un determinado defecto de un mecanismo de reparación no afecta de la misma forma a todas las células. En algunas ocasiones se produce enfermedad aún a pesar de que la maquinaria de reparación está intacta. En este caso la alteración afecta a genes encargados de regular la respuesta al daño en el genoma, por lo que no funcionaría el sistema de alerta o detección ante una anomalía. En este grupo podemos incluir a la ya citada proteína p53, cuyo gen aparece mutado en numerosos procesos tumorales.
La anemia de Fanconi es otra enfermedad relacionada con la reparación del material genético (puede estar producida por la alteración de varios loci diferentes), en este caso lo que se cree que falla es el sistema de detección o respuesta al daño en el ADN, ya que los individuos que la padecen parecen tener los sistemas de reparación intactos.
8.5 Telómeros y senescencia replicativa
A mediados del siglo pasado (1961) Hayflick and Moorehead observaron que fibroblastos y otras células humanas no tumorales en cultivo eran capaces de llevar a cabo un número finito de duplicaciones. Llegado este momento las células perdían su potencial replicativo y entraban en una fase de “senescencia replicativa”. Dicho estado no supone la muerte inmediata de la célula, más bien sería una situación similar a una fase G0 del ciclo celular. El número de duplicaciones que es capaz de realizar una determinada célula en cultivo es más o menos constante (siempre que las condiciones del cultivo no se modifiquen) y se conoce como límite de Hayflick. Hoy sabemos que este fenómeno se debe, en gran medida, a la pérdida de parte del material genético no codificante localizado en la región terminal de los cromosomas, que se conocen con el nombre de telómeros. Algunas evidencias experimentales ponen de manifiesto una posible relación entre longitud de telómeros y envejecimiento in vivo ( Smith y Pereira-Smith,1996).
Los telómeros son regiones terminales de los cromosomas lineales, en las células humanas se componen de secuencias cortas altamente repetitivas colocadas una a continuación de otra (en tandem) con tamaños de hasta 15.000 pares de bases. En humanos la secuencia que se repite es: TTAGGG. En los telómeros no se han identificado, hasta el momento, secuencias codificantes (con genes), pero se sabe que son fundamentales para proteger los extremos de los cromosomas de ataques enzimáticos (exonucleasas celulares) o modificaciones químicas (producidas entre otros por radicales libres). En este sentido los telómeros actuarían de secuencias protectoras, minimizando cualquier posible alteración sobre los extremos de los cromosomas. Además, durante la mayor parte del ciclo celular el ADN telomérico se encuentra altamente compactado constituyendo parte de la heterocromatina. En la estructuración de esta cromatina intervienen –además del ADN– proteínas que se asocian a éste y se cree que lo protegen de posibles alteraciones causadas por agentes mutagénicos.
8.5.1 Telomerasa y mantenimiento de los telómeros
En cada ciclo de división se pierden en cada telómero una media de 30-200 pares de bases debido al mecanismo de polimerización utilizado por la polimerasa (Figura 10), por esta razón se considera a los telómeros como posibles “relojes replicativos” que limitarían el número de duplicaciones que una célula puede llevar a cabo. Una vez rebasado un cierto número de replicaciones, durante las que los telómeros se habrían ido acortando por el propio mecanismo replicativo, se empezarían a perder genes situados en las regiones adyacentes a los telómeros.
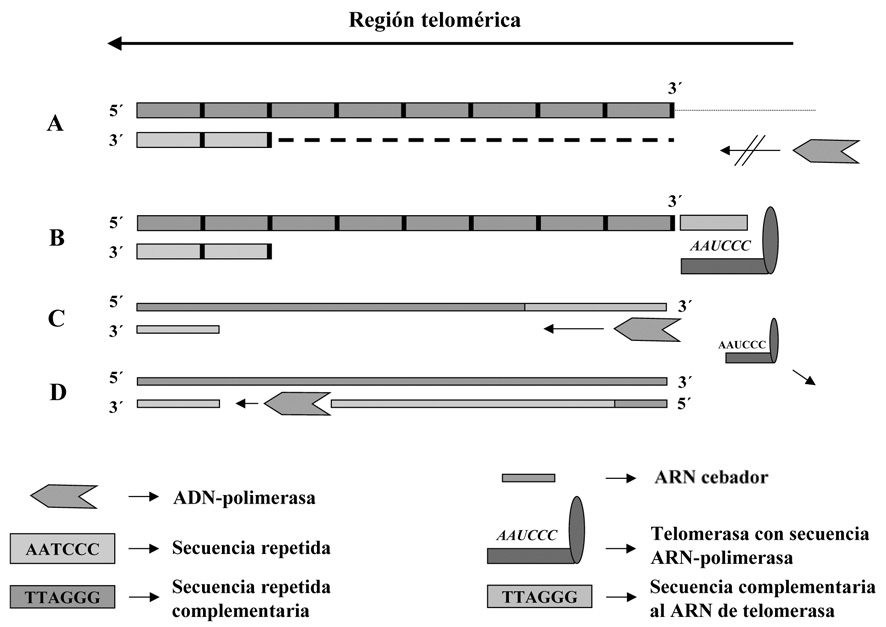
Figura 10. Mecanismo de acción de la telomerasa. En el esquema se representa la etapa la replicación del extremo de un telómero (los dibujos A y B tienen difrente escala que C y D).
-
A. La ADN polimerasa no puede replicar la hebra retardada por no encontrar un molde complementario en el punto de inicio, si la replicación terminase en estas condiciones la parte de cadena de ADN que no forma dúplex sería degradada perdiéndose esa parte del telómero.
-
B. La telomerasa sintetiza fragmentos de hebra molde aumentando su longitud hasta el punto en el que la polimerasa pueda iniciar una nueva replicativa.
-
C-D. La polimerasa inicia la síntesis de la hebra retardada a partir de un cebador y tomando como molde la hebra de síntesis directa previamente elongada por la telomerasa. Posteriormente la ligasa sellará la mella dejada por la polimerasa y se concluirá la síntesis de la hebra retardada.
La situación representada se refiere a la etapa final de la síntesis de un telómero, en ausencia de la telomerasa se perderían los fragmentos monocatenarios no replicados (la porción que está sobre la línea discontinua) cada vez que la célula salga de una fase S de su ciclo.
Dependiendo del número e importancia de los genes dañados por este mecanismo se vería afectada la viabilidad de la célula. No obstante, en líneas celulares destinadas al mantenimiento de estirpes celulares más diferenciadas (células germinales de tejidos reproductivos y no reporductivos), así como en líneas celulares inmortales (como las tumorales), el acortamiento telomérico es inexistente o está minimizado. Esta circunstancia se explica por la presencia de una ribozima (enzima con parte proteica y parte de ácido ribonucleico), con funciones de transcriptasa inversa llamada telomerasa, que actuaría de manera coordinada con la polimerasa del ADN, evitándose de esta forma la pérdida de material cromosomómico al final de cada ciclo replicativo (para comprender mejor la dinámica del fenómeno visitar la página: http://faculty.plattsburgh.edu/donald.slish/Telomerase.html).
La telomerasa está compuesta por una parte proteica y por una corta cadena de RNA (Figura 10), complementaria de la secuencia que se repite en los telómeros de los cromosomas de la célula que la produce. Los genes humanos que codifican tanto para la fracción proteica de la polimerasa, como para el pequeño fragmento de ARN se expresan activamente en células germinales, pero están silenciados o casi inactivos en la mayoría de las células diferenciadas. No obstante estos genes reactivan su expresión en la mayoría de las células cancerosas, donde la acción de la telomerasa es necesaria para sostener la alta tasa replicativa característica de las células que forman un tumor.
La relación entre telomerasa y senescencia replicativa se pudo comprobar in vitro al inmortalizar fibroblastos transfectados con los genes que codifican para la síntesis de la telomerasa. No obstante la relación causal entre telomerasa y envejecimiento está aún lejos de demostrarse de forma evidente, los ensayos diseñados al respecto han dado resultados contradictorios, además parece que la contribución real de la telomerasa in vivo al mantenimiento de los telómeros no parece ser tan evidente como se pensaba.
Los telómeros son muy sensibles a la presencia de radicales libres (Raha y Robinson, 2000; Eppel, 2004), los estudios encaminados a establecer la relación entre daño telomérico y estrés oxidativo ponen de manifiesto una clara relación causa efecto, observándose notables acortamientos teloméricos en células cultivadas en condiciones en las que aumenta la concentración intracelular neta de radicales del oxígeno. La forma en la que el ataque por radicales libres produce el acortamiento telomérico no se conoce con exactitud, pero podrían estar involucradas alteraciones tales como rotura de hebras y otras modificaciones químicas en nucleótidos situados en el extremo del cromosoma.
8.6 Conclusión
De todo lo anteriormente expuesto podemos concluir que, en mayor o menor medida, el envejecimiento de un organismo es debido a la senescencia o muerte de parte de sus células provocado por un deficiente mantenimiento de la copia del genoma.
La permanente agresión a la que se ve sometida la copia del ADN en las células de los diferentes tejidos a lo largo de la vida del individuo, genera una inestabilidad del genoma que se manifiesta en forma de mutaciones que provocan: productos deficientes, bloqueo de la transcripción o de la replicación, etc. Estas circunstancias son interpretadas por la célula como una situación de estrés al cual ha de responder de manera eficiente, si no quiere ver comprometida su viabilidad o funcionalidad, y por ello determina en gran medida el ritmo al que una célula envejece, ya que en él influyen dos elementos contrapuestos:
- A) La frecuencia con la que se producen daños en el genoma.
- B) La rapidez y eficiencia con que los mismos son detectados y reparados por la célula. Según esta hipótesis, aquellos individuos con una tara genética en la que se vean afectados loci que codifiquen para productos relacionados con sistemas de reparación del ADN, serían más susceptibles de presentar un envejecimiento prematuro.
En el plano experimental parece evidente la relación causa-efecto entre alteraciones genéticas que afectan a mecanismos de reparación del ADN y enfermedades asociadas al envejecimiento, tales como cáncer o síndromes progeroides. Por ello se espera que el estudio de dichos mecanismos nos permitirá determinar el impacto fenotípico de los daños acumulados a lo largo del tiempo en el genoma, y además contribuirá a establecer nuevas estrategias de prevención, determinación de susceptibilidad genética, diagnóstico y terapia racional.
En lo referente al papel biológico de la telomerasa su función como agente estabilizador de las regiones teloméricas parece claro. No obstante el mantenimiento de la integridad de los telómeros es complejo y en él intervienen otros muchos elementos además de la telomerasa. Dada la complejidad de este fenómeno, no podemos considerarlo como el único regulador de la senescencia replicativa. No hay que olvidar tampoco que la telomerasa juega un papel importante en el mantenimiento de la inmortalidad de las células cancerosas.
Referencias
- Ames B. N., Shigenaga M. K., Hagen T. M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA. 1993;90:79115-79122.
- Barnes D. E., Non-homologous end joining as a mechanism of DNA repair. Curr. Biol., 2001;11:R455-R457.
- Barnes D. E. DNA damage: air –breaks? Current Biology 2002;12:R262-R264.
- Eppel S., Blackburn E. H., Lin J., Dhabhar F. S., Adler N. E., Morrow J. D., Cawthon R. M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA. 2004;101,17312-17315.
- Halliwell B., Gutteridge J. M. C. Free radicals in biology and medicine. Halliwell B. and Gutteridge J. M. C (eds.) Oxford university press. Oxford.1999.
- Harman D. The aging process: Major risk factor for disease and death. Proc. Natl. Acad. Sci. USA. 1991;88:5360-5363.
- Hasty P. and Vijg J., Genomic Priorities in Aging. Science. 2002;296:1250-1251.
- Hoeijmakers J. H. Genome maintenance mechanisms for preventing cancer. Nature. 2001; 411:366-374.
- Hayflick L., Moorhead P. S. The serial cultivation of human diploid cell strains. Exp. Cell. Res. 1961;25:585-621.
- Johnson B. F., Sinclair D. A., Guarente L. Molecular Biology of Aging. Cell. 1999;96: 291-302.
- Klaunig J. E. and Kamendulis L. M. The role of oxidative stress in carcinogenesis. Ann. Rev. Pharmacol. Toxicol. 2003;44:239-267.
- Kolodner R. D. Guarding against mutation. Nature. 2000;407:687-688.
- Lombard D. B., Chua K. F., Mostoslavsky R., Franco S., Gostissa M., Alt. F. W. DNA repair, genome stability, and aging. Cell. 2005;120:497-512.
- Raha S., Robinson B. H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000;2510:502-508.
- Shiloh Y., Lehmann A. R. Maintaining integrity. Nat. Cell. Biol. 2004;10:923-928.
- Smith R. J. and Pereira-Smith O. M. Replicative senescence: Implications for in vivo aging and tumor suppression. Science. 1996;273:63-67.
- Watson J., Hopkins N. H., Robert J. W. y Steitz A. M. The mutability and repair of DNA, en: The molecular biology of the gene. Gillen J. R. (ed.) The Benjamin Cummings Pub. Co. Inc. Menlo Park, California. 1987. 339-357.
- Wood R. D. DNA repair in eukaryotes. Annu. Rev. Biochem. 1996;65:135-167.
- Wood R. D. Variants on a theme. Nature. 1999;399:639-640.
- Zglinicki T., Bürkle A., Kirkwood T. B. L. Stress, DNA damage an ageing, an integrative approach. Exptl. Geront. 2001;36:1049-1062.