• MC-F-015. Capítulo 15. Neurodegeneración y aportaciones terapéuticas.
Frente al proceso de envejecimiento neuronal que se podría llamar fisiológico o normal, ampliamente abordado en capítulos anteriores, destacan todos aquellos procesos que se caracterizan por la neurodegeneración patológica, cuyo sustrato neuropatológico es la atrofia neuronal acompañada de gliosis reactiva. Esta lesión fundamental se acompaña de alteraciones específicas en el parénquima cerebral que han de definir, por su ubicación y por su naturaleza, la enfermedad neurodegenerativa concreta. Las manifestaciones clínicas y la topografía de las lesiones predominantes son la base de clasificación de las principales enfermedades neurodegenerativas (Tabla 1).
De todas ellas, el presente capítulo se va a centrar en la enfermedad de Alzheimer porque es la expresión singular de un envejecimiento claramente patológico y que, además, presenta una importancia epidemiológica y una trascendencia social de enormes dimensiones, en una época en que la esperanza de vida se ha prolongado de manera tan vertiginosa y tan intensa. Junto a ella, estudiaremos un cuadro patológico genético especial, el síndrome de Down, porque ofrece también signos propios de un envejecimiento peculiar.
|
CLASIFICACIÓN DE LAS PRINCIPALES ENFERMEDADES NEURODEGENERATIVAS |
|
Síndromes en los que predomina la demencia
Enfermedades de los ganglios de la base
- Enfermedad de Parkinson. - Parkinsonismos plus: • Parálisis supranuclear progresiva. • Atrofias multisistémicas. • Otros.
- Enfermedad de Huntington. - Distonías de torsión y focales. - Síndrome de Gilles de la Tourette. - Temblor esencial. - Enfermedad de Hallervorden-Spatz.
Síndromes espinocerebelosos
Enfermedades de la motoneurona
- Atrofias musculares espinales. - Parálisis bulbares.
- Esclerosis lateral primaria.
Polineuropatías degenerativas |
Tabla 1. Tomada de Pascual, 2004.
15.1 La Enfermedad de Alzheimer
15.1.1 Características generales
La enfermedad de Alzheimer (EA) es una enfermedad neurodegenerativa que constituye la causa más frecuente de demencia. Su prevalencia se incrementa con la edad: es muy rara antes de los 40 años, aparece aproximadamente en el 3% de las personas mayores de 65 años y casi en el 50% de las mayores de 85 años. Aunque su etiología es desconocida, se conocen ya algunos factores que claramente están involucrados en su aparición, como se discutirá más adelante.
Se distinguen clásicamente dos formas de EA: la de origen y presentación familiar y la de aparición esporádica no familiar. En el primer tipo, el número de casos observados es menor del 1% del total, la enfermedad se inicia precozmente, entre los 40 y los 60 años y su origen es monogénico, debido a la mutación en tres genes: APP (proteína pre-amiloide) en el cromosoma 21, PSEN1 (presenilina 1) en el cromosoma 14 y PSEN2 (presenilina 2) en el cromosoma 1. Estas mutaciones poseen un alto grado de penetrancia y provocan un marcado aumento en la producción del péptido ß-amiloide (Aß42) y de su deposición en el cerebro, hecho que, como enseguida se explicará, caracteriza a la enfermedad de Alzheimer. Adicionalmente existe un cuarto gen, el APOE (alelo ε4) en el cromosoma 19, que es otro elemento de riesgo especialmente en las formas no familiares, tardías, de origen esporádico.
Estas formas tardías comienzan en edades por encima de los 60 años y son sin duda las más corrientes. Tienen un origen multigénico, multifactorial y complejo, en donde la presencia de varios factores de riesgo facilita el desarrollo de la enfermedad. En la actualidad se investiga intensamente para detectar otros genes de mucha menor penetrancia pero que constituyen factores de riesgo (Bertram y Tanzi, 2004). En el cromosoma 6, y en su región 6p21, se analizan diversos candidatos (HLA-A, HFE, TNFA, HAPA1B). En el cromosoma 10, en su región 10q24, se postulan varios genes entre los que destaca el IDE, que codifica la enzima que metaboliza la insulina y la proteína Aß en su forma monomérica. En el cromosoma 11, en su región 11q23, el gen BAC que codifica una ß-secretasa (véase más adelante el papel de las secretasas en la formación de ß-amiloide).
Macroscópicamente, la EA presenta una atrofia cerebral difusa que predomina en las circunvoluciones frontales, parietales y temporales mediales, con preservación relativa de las áreas motoras primaria, somatosensorial y visual. Las lesiones básicas se distribuyen por el neocórtex y el paleocórtex. El área entorrinal suele ser la que primero se afecta, lo que contribuye a la desaferenciación del hipocampo, y la que va a terminar gravemente afectada. También se degeneran los núcleos diencefálicos o infratentoriales de proyección neocortical, fundamentalmente los de naturaleza colinérgica (núcleo basal de Meynert, banda diagonal de Broca y núcleo septal medial), serotonérgica (algunos núcleos del rafe) y noradrenérgica (locus coeruleus).
Microscópicamente se aprecia una pérdida neuronal progresiva, en la que se aprecian fenómenos de apoptosis. Junto a ella, y probablemente como fenómenos causantes de esa pérdida, existen unas lesiones características que son los ovillos neurofibrilares y las placas seniles o neuríticas (Figura 1); pueden observarse también cuerpos de Lewy y de Hirano, degeneración granulovacuolar y depósitos amiloides en la pared de los vasos intracraneales. Estas lesiones suelen aparecer inicialmente en el territorio entorrinal (estadios I/II); posteriormente se suman las lesiones en hipocampo y amígdala (estadios III/IV), y por último se afectan las grandes áreas asociativas del neocórtex (estadios V/VI) (Braak y Braak, 1996).
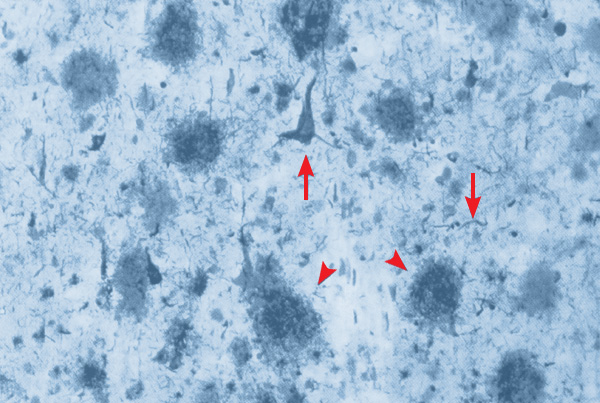
Figura 1. Neuropatología principal de la enfermedad de Alzheimer, visualizada al microscopio en corteza cerebral de un paciente. Se aprecian las placas de amiloide (puntas de flecha) y los ovillos neurofibrilares (flechas).
Los ovillos neurofibrilares son estructuras filamentosas argirófilas que se sitúan en el soma neuronal y las dendritas. Están formados por filamentos helicoidales emparejados de 10 μm de espesor cuya periodicidad es de 80 μm. Estos filamentos están compuestos fundamentalmente de proteínas tau (τ), unas proteínas que normalmente contribuyen a la estabilización de los microtúbulos pero que, cuando se encuentran hiperfosforiladas como es el caso de las que se encuentran en los ovillos neurofibrilares, disminuye su interacción con los microtúbulos y los desestabilizan. En consecuencia los microtúbulos se despolimerizan, las extensiones citoplásmicas se retraen, y se forman agregados aberrantes intensamente neurotóxicos. Las proteínas τ a su vez, pueden agregarse cuando están en altas concentraciones. Se desconoce la razón de esta hiperfosforilación de τ.
Las placas seniles tienen un diámetro entre 5 y 150 μm y están compuestas principalmente por un núcleo central de polímeros de un péptido denominado beta amiloide (Aß) y materiales inorgánicos, rodeado de neuritas en diverso grado de degeneración. Aß es un fragmento de la proteína pre-amiloide (APP), una proteína transmembrana que posee una región extracelular, otra intramembranal y otra citoplásmica. La biología molecular del eje APP → Aß se ha convertido en elemento sustancial de la patogenia de la enfermedad de Alzheimer por un hecho crítico: como se ha indicado anteriormente, ciertas mutaciones espontáneas en los cromosomas 21, 14 y 1 son causa de enfermedad de Alzheimer precoz de carácter hereditario, con abundante acumulación de Aß. Pues bien, algunas de estas mutaciones corresponden al gen de la APP situado en el brazo largo del cromosoma 21; y otras mutaciones se encuentran en los genes de la presenilina-1 (PS-1) y presenilina-2 (PS-2), dos proteínas que juegan un papel determinante en el procesamiento de la APP y están situados en los cromosomas 14 y 1, respectivamente. Es necesario, por tanto, prestar atención a la forma en que la proteína APP es procesada tanto en condiciones normales como patológicas. Esto obliga a centrar la atención en el procesamiento de la Aß porque ahí puede estar uno de los factores patogénicos más determinantes de la enfermedad de Alzheimer, y consiguientemente, se convierte en una de las dianas terapéuticas más incitantes. Es lo que se denomina la cascada amiloide (Figuras 2 y 3).
15.1.2 El papel del péptido Aß y la cascada amiloide
La APP es fisiológicamente seccionada por una α-secretasa, que es una metalo proteasa asociada a membrana. Produce α-APPs y un fragmento C-terminal asociado a membrana con 83 residuos, el C83, por cortar en Lys 686-Leu 689. Este C83 es también sustrato para la γ-secretasa, dando origen a la formación de p3 que es una forma truncada N-terminal de 3kDa a partir del C83 (Figura 2).
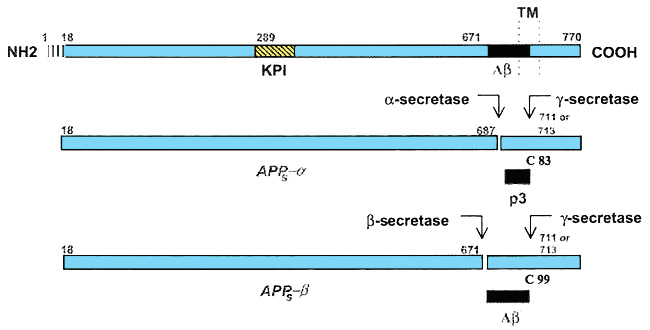
Figura 2. Visión esquemática de la proteína APP y de sus principales derivados mediante procesamiento de las secretasas. KPI: dominio tipo Kunitz que contiene el inhibidor de una serina-proteasa. TM: segmento transmembranal de la célula. Véase el texto. (Tomada de Selkoe y Kopan, 2003).
La proteína Aß no sólo se produce en el cerebro en condiciones patológicas; se forma en prácticamente todos los tipos de células y se incrementa su producción si se transfecta APP en líneas celulares inmortalizadas. Para ello debe actuar primero la ß-secretasa: genera la porción N-terminal de la Aß, rompiendo la APP en su sección luminal/extracelular, a unos 30 residuos del dominio transmembrana, originando ß-APP y un fragmento C-terminal de 99 residuos asociado a la membrana, el C-99. Posteriormente actúa la γ-secretasa que hidroliza el residuo en medio del dominio transmembrana para originar la proteína Aß que suele tener 42 residuos, por lo que suele llamarse Aß42 (Figura 2). Aunque la mayoría de los tipos celulares producen Aß, lo cual significa que la ß-secretasa se encuentra ampliamente extendida, es cierto que se genera más Aß en cultivos primarios de cerebro que en los de células periféricas, y las neuronas muestran más actividad ß-secretasa que los astrocitos (quizá esto explique en parte por qué Aß se agrega de forma selectiva en el cerebro). Los niveles de ARNm son mayores también en las neuronas que en la glía.
La ß-secretasa (ß-site APP Cleaving Enzyme o BACE1) es una aspartil proteasa de 501 aminoácidos que contiene un único dominio transmembrana cerca de C-terminal. Necesita tener 2 aspartatos, en D 93 y D 289, para mostrar actividad. La proteasa que la activa es una convertasa tipo furina. A pesar de ser una aspartil proteasa, su actividad no es inhibida por un inhibidor de aspartil proteasas de amplio espectro, la pepstatina A. Pero la incorporación de estatina a sustratos peptídicos contribuye a formar potentes inhibidores. El gen de la ß-secretasa (BACE1) está en el cromosoma 11, pero no se ha detectado todavía ninguna mutación originadora de la EA familiar en este gen.
La ß-secretasa parece ser similar en su modo de actuar a otras “sheddasas” asociadas a membrana como son la TNF-α secretasa, la ACE secretasa, la TGF-ß secretasa. La ß-secretasa corta en la secuencia EVKM*DAEF; el principal sitio de ruptura es en Asp 1, pero puede hacerlo también en Glu 11.
Diversas mutaciones del gen APP son capaces de producir enfermedad de Alzheimer, así como la forma holandesa de angiopatía amiloide. La mutación en la posición 717 (14-16), que reemplaza valina por fenilalanina, isoleucina o glicina justo tras el extremo C-terminal del péptido Aß, favorece la formación de fragmentos de mayor longitud como el Aß42. La doble mutación 670/671, en el extremo N-terminal, favorece la acción de la ß-secretasa aumentando la producción de péptido Aß. Por último, la mutación responsable de la forma holandesa de angiopatía amiloide en el residuo 693 produce un péptido ß con mayor estabilidad al agregarse.
También el exceso de APP por sobredosis génica, como ocurre en el síndrome de Down o trisomía del cromosoma 21, provoca la aparición precoz de Aß42 y demás hallazgos neuropatológicos similares a los de la enfermedad de Alzheimer, como más adelante se explicará.
Las presenilinas 1 y 2 (PS1 y PS2, respectivamente) fueron identificadas en 1995, en una búsqueda por encontrar los genes causantes de formas familiares de enfermedad de Alzheimer ligadas a los cromosomas 14 y 1. Pronto se comprobó la extraordinaria asociación entre PS y enfermedad de Alzheimer, ya que numerosos casos de EA familiar se debían a mutaciones originadas en los genes de dichas proteínas. Las dos presenilinas son proteínas de membrana que poseen 8 segmentos transmembrana (TM) y presentan una homología entre ellas del 65%.
El gen de la PS1, como ya se ha comentado, se encuentra en el cromosoma 14. Se han descrito 64 mutaciones que inducen enfermedad de Alzheimer familiar. El gen de la PS2 se encuentra en el cromosoma 1. Se han descrito 6 mutaciones que inducen enfermedad de Alzheimer familiar, y algunas de ellas la originan a los 25 años. Todas las mutaciones originan aumentos específicos de Aß42, demostrables en células transfectadas, en ratones transgénicos, en plasma sanguíneo y en medios de cultivos de fibroblastos.
Las presenilinas modulan la actividad de la γ-secretasa para incrementar la ruptura del enlace Ala 42-Thr 43. ¿Podrían ser las presenilinas las γ-secretasas? Es muy posible que las presenilinas contengan el sitio activo de la γ-secretasa (Selkoe y Kopan, 2003). En el caso de las mutaciones de los genes PS1 y PS2 se facilita el corte de la APP por parte de las ß-secretasa y γ-secretasa (Figura 3).
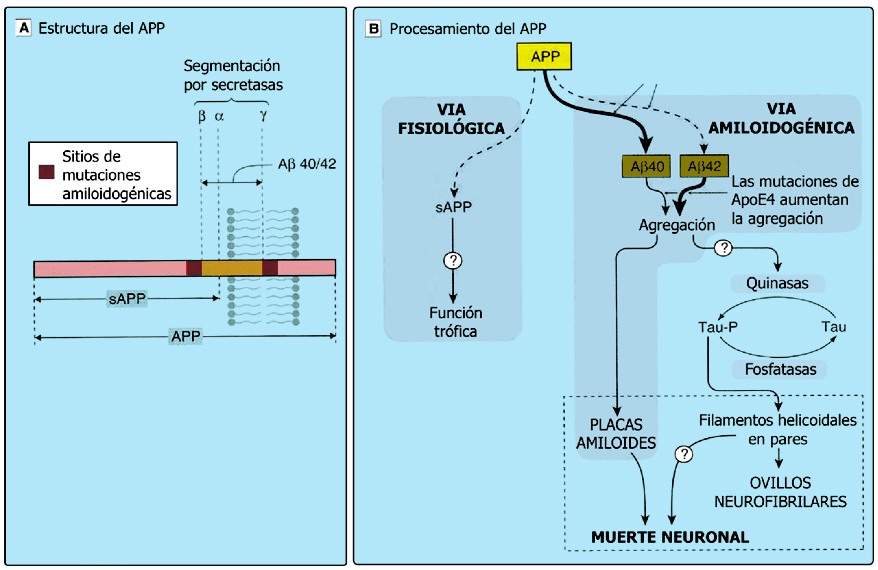
Figura 3. Cascada de la producción de ß-amiloide a partir de la proteína precursora APP. En (A) se muestran los sitios de segmentación de la APP por parte de las secretasas. En (B) se muestra la vía de procesamiento patológico por acción de las ß- y γ-secretasas, para originar los péptidos amiloides Aß42 y Aß40. (Tomada de Rang et al., 2004).
Existe otro factor de riesgo, aunque no sea de modo determinante como es el caso de las mutaciones antes señaladas, para la aparición de enfermedad de Alzheimer: la presencia del alelo ε4 de la apolipoproteína E (apoE, cuyo gen se localiza en el cromosoma 19). El alelo ε4 afecta al procesamiento de Aß: su presencia parece incrementar la deposición de las proteínas Aß40 y Aß42 en los cerebros (McNamara et al. 1998). Se desconoce todavía el mecanismo por el que el alelo ε4 de la apoE facilita la mayor presencia y deposición del amiloide Aß42; si se trata de una mayor velocidad en la formación de fibrillas del amiloide, o de una menor velocidad en la limpieza y desaparición de dichas fibrillas. Pero lo más interesante es la demostración en cultivos de células musculares lisas de que la presencia concurrente de estrés oxidativo favorece la acumulación de apoE4 y la formación de depósitos apoE-Aß.
El péptido Aß es fácilmente reconocible por su capacidad para autoagregarse y formar amiloide. El amiloide consta de grandes fibrillas cuya estructura está en conformación ß. Los péptidos Aß constan de 39-43 residuos; en las placas seniles abundan las especies más hidrofóbicas de 42-43 residuos, que fácilmente forman las fibrillas.
Muchos estudios apoyan la hipótesis de que las fibrillas inducen la neurodegeneración propia de la EA: es la hipótesis de la cascada de amiloide (Figura 3). Si se exponen neuronas cultivadas a Aß sintético aparece muerte celular. Estas soluciones tóxicas contienen abundantes fibrillas de amiloide, pero además contienen especies más pequeñas.
Hay muchos datos en la literatura que favorecen la asociación entre la formación de Aß42 y EA. El problema que más se debate, el problema de fondo, es saber si la toxicidad se debe a la formación de fibrillas de Aß o si se debe a Aß42 pero en una forma previa a la formación de fibrillas (Klein et al., 2001). De hecho existe una pobre correlación entre la carga de amiloide fibrilar y el grado de disfunción neurológica. Los depósitos de amiloide pueden estar distantes de las zonas con pérdida neuronal. El mejor índice patológico de demencia es la pérdida de terminaciones sinápticas, y esto se correlaciona pobremente con la carga de amiloide.
Si las manifestaciones de la enfermedad guardan una relación muy débil con la carga de amiloide, ¿cuál es, entonces, el papel que desempeña la Aß? La solución puede ser la siguiente:
Las fibrillas de Aß pueden no ser las únicas formas neurotóxicas para la EA, y quizá ni siquiera las más importantes. Existen varias formas de ensamblaje de la Aß:
- El monómero Aß es inocuo; tiene que autoasociarse para hacerse neurotóxico, sin necesidad de hacerse fibrilar.
- Protofibrillas: estructuras curvilíneas de <200 μm de longitud y 4-11 μm de diámetro. Son neurotóxicas, producen estrés oxidativo y muerte neuronal.
- Oligómeros pequeños: más estables; aparecen sin necesidad de convertirse en fibrillas. Aumentan en células transfectadas con presenilinas mutadas de EA familiar. Son los denominados Aß-derived diffusible ligands o ADDLs. Estas formas solubles pueden afectar a las neuronas, pero se escapan de la detección en las mediciones de amiloide sólido. Los ADDLs muestran neurotoxicidad regional selectiva, en CA1 de hipocampo y en corteza entorrinal, pero no en cerebelo, y producen con gran rapidez (en menos de 1 hora) inhibición completa de la potenciación a largo plazo (LTP) en hipocampo, lo que puede estar relacionado con la perturbación de la memoria. Estas toxinas solubles serían las responsables de la pobre correlación entre el amiloide fibrilar y la progresión de la enfermedad, y ofrecerían una explicación unificadora de la patogenia de la EA. De hecho, la pérdida de sinapsis en la EA correlaciona con el Aß soluble, pero no con el amiloide (Lue et al., 1999). Cinco grupos de investigadores han encontrado que los cerebros de EA contienen oligómeros Aß.
El estudio de modelos animales resulta esclarecedor. Ratones transgénicos con APP humano contienen niveles elevados de Aß y múltiples déficits neurológicos, pero no contienen depósitos de amiloide. Es decir, estos modelos animales vienen a ser como una repetición en exagerado de la débil correlación entre amiloide y enfermedad humana.
De hecho, hay ratones transgénicos con APP humano que muestran neurotoxicidad (pérdida de sinapsis) y no presentan depósitos de amiloide. Transgénicos con APP mutante presentan también pérdida de sinapsis aunque también depósitos de amiloide. No hay, pues, correlación entre depósitos y pérdida sináptica. En cambio, hay correlación entre pérdida sináptica y niveles de Aß soluble. Por consiguiente, la patología del SNC independiente de la formación de placas puede ser explicada por la neurotoxicidad de los oligómeros Aß.
En consecuencia, las toxinas solubles derivadas de Aß podrían convertirse en el blanco o diana más crucial en el desarrollo de la vacuna para la EA. La respuesta final en relación con la naturaleza de las toxinas Aß en la EA podría depender de los anticuerpos que marcan epitopos específicos de particulares especies derivadas de Aß. Lo cual tiene aún mayor interés en vista de los efectos terapéuticos observados con los anticuerpos Aß. ¿Cuál será la mejor diana para una óptima vacuna en la EA?
15.1.3 Modelos animales de EA
Se han generado animales transgénicos que expresan mutaciones en APP, PS1 y PS2, y animales que expresan más de una de estas mutaciones. En los modelos APP de ratón se aprecian placas neuríticas con acumulaciones de Aß relacionadas con la edad en el hipocampo y en la corteza cerebral, con activación de astrocitos y células microgliales en las regiones en donde se encuentran las placas, y con degeneración de las terminaciones colinérgicas. Pero ni en los modelos APP ni en los PS se observan los ovillos neurofibrilares, ni una sustancial pérdida de neuronas en la corteza o en los núcleos colinérgicos basales o en el locus coeruleus. En los ratones que expresan proteínas τ mutantes se observan los ovillos neurofibrilares, y éstos aumentan si los ratones expresan además APP mutante (German y Eisch, 2004).
15.1.4 Posibilidades terapéuticas
A la vista de lo expuesto sobre la patogenia de la enfermedad de Alzheimer, podemos plantear las siguientes posibles dianas terapéuticas:
- A) Impedir la formación del depósito de la proteína ß-amiloide (Aß), o facilitar su aclaramiento.
- B) Anular la hiperfosforilación anormal de proteína tau.
- C) Combatir la neuroinflamación.
- D) Corregir el desequilibrio de neurotrofinas.
- E) Modificar el ambiente de la neurotransmisión química.
- F) Contrarrestar las consecuencias del estrés oxidativo.
- G) Terapia génica.
- H) Otros recursos: estatinas, Ginkgo biloba, etc.
No es objeto de esta exposición revisar cada una de las posibles actuaciones terapéuticas. En la actualidad, y dentro de sus evidentes limitaciones, el incremento de la actividad colinérgica con los inhibidores de la acetil colinesterasa y de la actividad glutamatérgica con memantina son los únicos y endebles instrumentos de que disponemos (Flórez, 2003b). Visto el papel preponderante de la cascada amiloide, se está realizando un notable esfuerzo investigador por contrarrestarla, bien mediante inhibición de las secretasas, bien por anulación inmunitaria de la Aß.
15.2 El Síndrome de Down
Son varias las razones que justifican que abordemos el Síndrome de Down (SD) en el contexto de este libro, justo en el final de su exposición. En primer lugar, en este síndrome existe un proceso claro de envejecimiento precoz cuyo análisis nos puede ayudar a resaltar algunos de los conceptos que se han ido explicando a lo largo de esta obra. En segundo lugar, ha habido una revolución sustancial en el modo de contemplar y abordar la atención a las personas con SD, de modo que cada vez las vemos más integradas y autónomas en todos los ámbitos de nuestra sociedad (Flórez, 2003a). En tercer lugar, la atención médica ha prolongado notablemente la esperanza de vida de estas personas, hasta alcanzar una media próxima a los 60 años. Por último, el análisis del proceso de envejecimiento en el SD, como modelo natural humano, nos puede ayudar a encontrar soluciones terapéuticas para otros tipos o formas de envejecimiento.
15.2.1 La naturaleza del Síndrome de Down
El SD o trisomía 21 es una entidad que en la actualidad constituye la causa genética más frecuente de discapacidad intelectual y malformaciones congénitas. Es el resultado de una anomalía cromosómica por la que los núcleos de las células del organismo humano poseen 47 cromosomas en lugar de 46, perteneciendo el cromosoma excedente o extra al par 21. Como consecuencia de esta alteración, existe un fuerte incremento en las copias de genes del cromosoma 21, lo que origina una perturbación en el programa de expresión de muy diversos genes, no sólo del cromosoma 21 sino de otros cromosomas. Este desequilibrio génico ocasiona modificaciones en el desarrollo y función de los órganos y sistemas, tanto en las etapas prenatales como postnatales. Consiguientemente, aparecen anomalías visibles y diagnosticables; unas son congénitas y otras pueden aparecer a lo largo de la vida. El sistema más comúnmente afectado es el sistema nervioso y dentro de él, el cerebro y cerebelo; por este motivo, casi de manera constante la persona con Síndrome de Down presenta, en grado variable, discapacidad intelectual.
Pese a la existencia común y constante de los tres cromosomas 21, el modo en que se desarrolla la acción de sus genes –lo que denominamos su expresión génica– varía en cada individuo. Por este motivo, el grado de afectación de los distintos órganos y sistemas es extraordinariamente variable. Esto hace que el número y la intensidad de las alteraciones orgánicas propias de cada persona pueda ser muy diferente. Esta realidad incluye a la discapacidad intelectual; al ser consecuencia de la patología cerebral derivada del desequilibrio en la expresión génica, las variaciones individuales de esta patología repercutirán en el grado y la manifestación de la discapacidad que, por tanto, habrá de ser considerada, evaluada y tratada de manera individual. Pero como la propia realidad cerebral y sus consecuencias –la personalidad, la inteligencia, la capacidad adaptativa– están fuertemente condicionadas por el influjo ambiental –educación, nutrición, bienestar–, y éste es también altamente variado para cada persona, el resultado final del funcionamiento vital del individuo con SD es una condición que no es predecible en su inicio y es ampliamente influenciable en su desarrollo (Flórez y Ruiz, 2003).
Por otra parte, el desequilibrio génico opera sobre los órganos de forma altamente independiente. Esto significa, en primer lugar, que distintos individuos presentan distintas alteraciones orgánicas; y en segundo lugar, que la intensidad de la alteración en un órgano puede ser muy diferente de la que ocurra en otro órgano. Y aun dentro de un mismo órgano complejo como es el cerebro, la alteración puede diferir notablemente de unas áreas y núcleos a otras. Como ejemplo, el hecho de que el corazón pueda estar muy afectado no significa que el cerebro lo haya de estar en el mismo grado; o que rasgos faciales muy característicos signifiquen grave afectación del cerebro. Puede ocurrir, sin embargo, que la mala función de ciertos órganos vitales –por ejemplo, corazón, tiroides– limite la actividad del individuo y condicione negativamente el desarrollo del cerebro y de sus funciones.
Los actuales programas de salud y educación han conseguido que la mayoría de las personas con Síndrome de Down alcancen un notable grado de integración escolar, laboral y social, así como de participación en las múltiples actividades sociales.
15.2.2 El envejecimiento en el Síndrome de Down
De acuerdo con nuestro actual estado de conocimientos, debemos partir de los siguientes datos (Flórez, 2002):
- La esperanza media de vida de las personas con SD se aproxima a los 60 años. Esta cifra es claramente inferior a la que ofrece la población general y la población con discapacidad intelectual que no tiene SD. La cifra es una media y su intervalo es muy amplio. Son varios los informes que van apareciendo sobre personas con SD que han llegado a la década de los setenta e incluso ochenta sin signos de demencia (Chicoine y McGuire, 1997).
- En cualquier caso, el SD es un claro factor de riesgo en lo que a la anticipación del envejecimiento y mortalidad se refiere. En el SD existe precocidad de envejecimiento.
- La tasa de aparición de EA en la población con SD es muy superior a la del resto de la población, incluida la que tiene discapacidad intelectual sin dicho síndrome. Además, la edad a la que se presenta EA en las personas con SD suele ser relativamente precoz, con una evolución media de 4-5 años.
- La aparición y la evolución de EA muestran un alto grado de variabilidad interindividual. De hecho, hay personas con SD que no han desarrollado demencia a edades muy avanzadas.
- Se estima que el 25% pueden mostrar signos y síntomas de demencia tipo Alzheimer a partir de los 35-40 años. El porcentaje aumenta con la edad, de modo que en la década de los sesenta el porcentaje alcanza cifras que son muy variables según las diversas estadísticas: entre el 30 y el 75%.
- Envejecimiento precoz en el SD no es sinónimo de EA. Se hace preciso distinguir entre lo que es un deterioro o declive (que incluye lo cognitivo) relacionado con la edad en el SD, y lo que es la instauración y presencia de demencia. De la misma manera, es necesario diferenciar muy bien a la demencia de la sintomatología que ofrecen otros procesos que pueden aparecer en el adulto con SD, como son la depresión, el hipotiroidismo o las pérdidas sensoriales, en especial la visión y la audición.
- La precocidad del envejecimiento de las personas con SD es una realidad. Entre sus manifestaciones destacan:
- A) La disfunción inmunitaria.
- B) La formación de cataratas.
- C) La presbiacusia.
- D) La atrofia de la piel.
- E) El declive en algunas habilidades cognitivas.
- F) La neuropatología tipo Alzheimer (sin demencia).
- G) La aparición plena de la EA.
Centraré mi análisis en la presentación de los datos biológicos que pueden explicar este proceso de envejecimiento precoz en el SD, por una parte, y esta tendencia marcada hacia la aparición de la EA, por otra.
15.2.2.1 El envejecimiento precoz: razones biológicas
Parece lógico partir de la base de que las peculiaridades características del envejecimiento en el SD son consecuencia directa de su particular dotación cromosómica: la sobredosis génica que aporta la presencia de un cromosoma extra del par 21. Hoy día vamos conociendo cada vez mejor que sobredosis de gen no significa necesariamente que vaya a haber sobreexpresión de ese gen en forma de una mayor cantidad de la proteína derivada de ese gen. Puede haberla o no; e incluso puede haber disminución de la expresión de otras proteínas (Engidawork y Lubec, 2003).
Numerosos datos experimentales indican que determinadas células del organismo con SD muestran signos claros de estrés oxidativo. El estrés oxidativo viene definido por la aparición y acumulación de un conjunto de productos derivados del metabolismo oxidativo, que ejercen una acción tóxica sobre las estructuras de la célula en donde se forman. La acumulación de estos productos se debe a que hay un exceso de producción, no compensada por la adecuada capacidad de neutralizarlos.
Los productos son los llamados radicales de oxígeno, que poseen una alta capacidad reactiva, es decir, tienen una gran capacidad de interactuar con otras moléculas de las estructuras celulares, modificándolas y, por así decir, debilitándolas para ejercer su correcta función. Con lo cual, a la larga, la célula va perdiendo su capacidad funcional e, incluso, puede llegar a morir.
Los principales radicales de oxígeno, que son generados como subproductos de la fosforilación oxidativa, son los siguientes:
- Aniones superóxido (•O2-).
- Radicales hidroxilo (•OH) e hidroperóxidos (-ROO•).
- Peróxido de hidrógeno (H2O2).
Las mitocondrias representan la principal fuente de producción de superóxidos en la mayoría de las células, y los aniones superóxido funcionan como precursores directos en la formación de peróxido de hidrógeno. El superóxido se forma dentro de las mitocondrias cuando los electrones son liberados fuera de la secuencia en la cadena de transporte electrónico, siendo transferidos directamente al oxígeno molecular.
Pues bien, en el SD hay una sobreexpresión del gen SOD1 que se encuentra en el brazo largo del cromosoma 21 (Figura 4), responsable de la formación de una enzima: la superóxido dismutasa 1. Esta enzima se encarga de canalizar ciertos radicales libres de oxígeno hacia la formación del peróxido de hidrógeno, del cual derivarán otros radicales de oxígeno que, en exceso, pueden ejercer una acción tóxica (Figura 5). En condiciones normales, la producción de estos radicales puede verse neutralizada por la acción de otras enzimas, la catalasa y la glutatión peroxidasa. Pero si hay sobreproducción de peróxido de hidrógeno por una actividad excesiva de la SOD1, o si la glutatión peroxidasa no contrarresta suficientemente el formado, habrá un exceso de radicales libres de oxígenos altamente reactivos que producirán peroxidación de lípidos de membrana y otras acciones hasta lesionar las estructuras celulares.
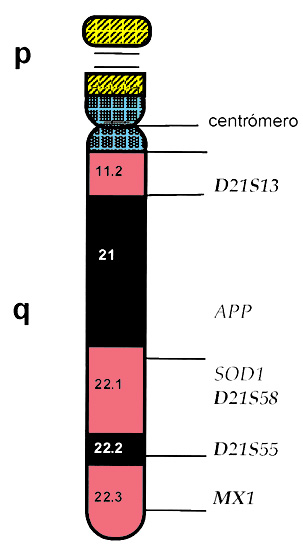
Figura 4. El cromosoma 21 humano mostrando la ubicación de los genes APP y SOD1.
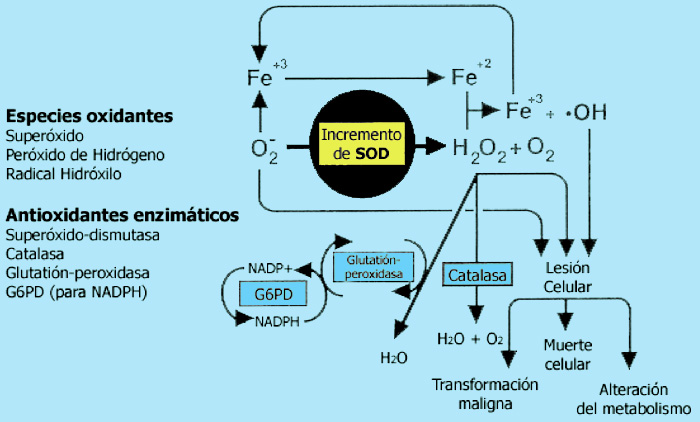
Figura 5. Mecanismos oxidativos en los que interviene la superóxido dismutasa (SOD1).
Como consecuencia del exceso de actividad de SOD1 en el SD, no acompañada por una mayor actividad de la catalasa y glutatión peroxidasa, se ha podido demostrar un incremento de la producción de superóxidos (aniones hidroxilo) en las mitocondrias de diversas células de las personas con SD. El resultado es que estas mitocondrias son menos eficientes con el tiempo. Aparecen lesiones o cambios conformacionales en sus lípidos, en sus proteínas y, lo que es más grave, en el propio ADN mitocondrial. La lesión de las proteínas de la cadena de transporte de electrones dentro de la membrana mitocondrial interna puede alterar la eficiencia de la cadena de transporte de electrones y, consiguientemente, el metabolismo oxidativo. Pero, además, la lesión del ADN mitocondrial provoca cambios más sustanciales; en efecto, a diferencia del ADN nuclear, el mitocondrial no se encuentra protegido por proteínas del tipo de las histonas; es decir, se encuentra más vulnerable frente a la lesión oxidativa, lo que termina por producir mutaciones o pérdida de bases en sus nucleótidos; y además, está menos sometido a la acción de procesos reparadores intrínsecos.
Todo este conjunto de alteraciones es lo que llamamos estrés oxidativo. Lo que nos interesa señalar aquí es que se han observado signos de estrés oxidativo ya durante el desarrollo fetal y etapa postnatal temprana en las neuronas de fetos y niños con SD. Ello, lógicamente, puede afectar negativamente a dos tipos de procesos:
- A) Los procesos de neoformación y, sobre todo, de diferenciación de las neuronas durante el desarrollo del sistema nervioso fetal, cuando las neuronas de diversas estirpes van madurando, migrando y ubicándose en sus diversos espacios y sitios, para originar las ricas redes interneuronales del cerebro.
- B) Los procesos de supervivencia neuronal, sobre todo en situaciones marcadas por el riesgo.
Un ejemplo que explica de forma muy gráfica la trascendencia del estrés oxidativo para la supervivencia y el desarrollo de las neuronas en el SD, viene revelado por los resultados que Busciglio y Yankner obtuvieron y publicaron en 1995. Estos investigadores extrajeron neuronas vivas de la corteza cerebral de fetos con SD (neuronas SD) y sin SD (neuronas no-SD) recién abortados (entre 17 y 21 semanas de gestación), y las mantuvieron vivas en cultivo durante una serie de días. Observaron que estas neuronas eran capaces no sólo de sobrevivir sino de desarrollar sus típicas prolongaciones y arborizaciones, y de establecer conexiones unas con otras. Pero había una importante diferencia: las neuronas no-SD se mantenían vivas y activas en el cultivo durante al menos 14 días, mientras que las neuronas SD sólo duraban 7 días: perdían sus conexiones y morían. En esas circunstancias, los investigadores estudiaron el metabolismo oxidativo, y comprobaron que las neuronas SD mostraban un importante incremento de las especies de oxígeno reactivo (3 a 4 veces mayor que en las neuronas no-SD) así como un aumento en el nivel de peroxidación de lípidos resultante de lo anterior. Más aún, la adición al líquido de cultivo de productos antioxidantes contrarrestaba parcialmente esos incrementos en las neuronas SD y restablecía su supervivencia.
Más recientemente, investigadores de ese mismo laboratorio han expuesto nuevas contribuciones. En este caso, utilizando la misma técnica de cultivo de neuronas fetales con y sin SD, analizaron la respuesta de las neuronas frente a un radical superóxido, el peróxido de hidrógeno (H2O2). El superóxido produjo notable reducción de la supervivencia neuronal por degeneración de las estructuras en ambos tipos de neuronas, con producción de radicales libres de oxígeno y aumento de peroxidación de lípidos. Es evidente, pues, que el peróxido de hidrógeno (que en el SD puede estar aumentado por el incremento de la dosis génica de SOD1) se convierte en un elemento desestabilizador de las neuronas SD. Estas neuronas muestran signos de estrés oxidativo, con consecuencias perjudiciales para ellas.
El fenómeno no es exclusivo de las células neuronales. Recientemente ha sido observado también, por ejemplo, en los macrófagos obtenidos de la sangre de niños con SD (1 a 8 años), en donde se apreció un aumento en la producción de superóxidos a nivel mitocondrial (Capone et al., 2002) y en cultivos de fibroblastos (Busciglio et al., 2002). Se aprecian alteraciones en las proteínas mitocondriales de personas con SD (Kim et al., 2000).
La conclusión que podemos obtener a partir de todos estos estudios es que ciertas células de las personas con SD están bajo la amenaza del estrés oxidativo, posiblemente expresado por la acumulación de radicales hidroxilo que reducen la capacidad funcional de la mitocondria. La lesión que estos radicales ejercen sobre los lípidos mitocondriales, las proteínas y el ADN mitocondrial pueden explicar la acción permanente y perjudicial que va erosionando la vida y la permanencia de las células, llevándolas hacia el envejecimiento y la muerte. Es así como explicaríamos el envejecimiento precoz, al que consideramos como una de las características más acusadas (aunque no constantes) del SD.
15.2.2.2 La neuropatología tipo-Alzheimer en el Síndrome de Down
Analicemos ahora la presencia de la neuropatología tipo-Alzheimer en el SD y las razones posibles de su presencia.
Se aprecia un aumento muy precoz en la formación de la APP, con sustanciales incrementos tanto en el plasma sanguíneo como en el cerebro de las personas con SD; y, además, a edades muy tempranas, muy anteriores a lo que se ve en el resto de la población. Esto es lógico, porque si en el cromosoma 21 está el gen APP responsable de la síntesis de esta proteína (Figura 4), la triple dosis de gen (por causa de la trisomía) puede producir mayor cantidad de proteína.
Pero una cosa es que haya mayor cantidad de proteína APP, y otra que haya una derivación de su procesamiento hacia la síntesis de la proteína neurotóxica, la Aß42, que es lo que ocurre en el SD. De hecho, es posible observar aumentos de los niveles plasmáticos de Aß42 en personas con SD (Tokuda et al., 1997; Mehta et al., 1998; Schupf et al., 2001), así como Aß42 soluble en los cerebros, en edades muy precoces e incluso prenatales (Teller et al., 1996; Lemere et al., 1996; Mori et al., 2002). Este Aß42 tan precozmente detectable se aprecia primero en localización intracelular; sólo después pasará al exterior y se agregará en placas amiloides. Tal presencia precoz de Aß42 no es detectable en cerebros normales.
15.5.2.2.1 Patogenia de Aß en el Síndrome de Down
En los cerebros jóvenes con SD (por debajo de los 30 años) se aprecia la acumulación de Aß en forma de depósitos difusos, todavía no asociados a degeneración neurítica. Estas placas difusas son tioflavina-S negativas, lo que indica ausencia de amiloide de tipo fibrilar. Las formas principales de Aß son mayoritariamente Aß42 y parcialmente Aß40. Con el tiempo, la forma Aß42 extracelular en el SD puede ser modificada post-translacionalmente por isomerización, racemización y oxidación.
A pesar de que exista una amplia acumulación de Aß extracelular tanto en el SD como en la EA de la población general, no se ha podido comprobar una clara asociación entre la presencia de este Aß y la demencia. De hecho, los ratones transgénicos que sobreexpresan APP pueden mostrar algunos signos de disfunción cognitiva antes de que se acumule el Aß insoluble y extracelular. Se piensa actualmente que no es esta Aß la forma crítica que condiciona la neurotoxicidad sino otra que es anterior a ella, se acumula antes y es probablemente más tóxica, a saber, las formas solubles que comprenden los llamados oligómeros de Aß, protofibrillas y otros ligandos difusibles que derivan de Aß, tal como se ha explicado anteriormente (Klein et al., 2001; Caughey y Lansbury, 2003; Lott y Head, 2005). Los oligómeros pueden incluso perturbar la función neuronal aun antes de provocar pérdida neuronal. Desconocemos la evolución temporal de esta acumulación de oligómeros Aß en el SD, que puede ser anterior o en paralelo con la de las formas de Aß intracelular y extracelular. Hay una pregunta crítica que hacer: ¿basta el incremento de APP en el SD (como consecuencia de la sobreexpresión del gen presente en el cromosoma 21) para que se produzca el exceso de Aß patológico, o hacen falta otros factores condicionantes que favorezcan el procesamiento del APP en esa dirección? Parece necesario que existan estos otros factores. Lo que resulta lógico es pensar que algunos de dichos factores condicionantes son aportados también por la propia trisomía 21, ya que es en ella en donde se aprecia la evolución frecuente hacia la neuropatología propia de la EA.
Dentro de las neuronas, el procesamiento del APP que da origen a las distintas especies de Aß tiene lugar en el sistema endosómico/lisosómico. Ciertamente, en el SD se aprecia siempre esa localización, junto con un aumento de la actividad endosómica. No basta que haya aumento de APP para que aparezca hiperactividad endosómica, hace falta algún factor adicional. De hecho, cuando un ratón sobreexpresa sólo APP no muestra ese patrón endosómico, y sí lo hace, en cambio, el ratón Ts65Dn que presenta una trisomía parcial de su cromosoma 16, el cual contiene una extensa región de genes sinténicos con los del cromosoma 21 humano; es decir, junto a la sobreexpresión del gen APP en este ratón trisómico hay también la de otros muchos genes (Cataldo et al., 2003).
¿Cuál puede ser, pues, el mecanismo responsable de que en el SD la APP producida en exceso derive su procesamiento hacia la patológica formación de Aß42? Ésta es la gran cuestión que se está intentando resolver.
15.2.2.2.2 Estrés oxidativo y formación de amiloide
De entre las diversas hipótesis, destacaré una que está siendo crecientemente aceptada y que empieza a verse apoyada por recientes datos experimentales. La hipótesis afirma que el factor responsable de la activación protagonizada por la acción de la ß-secretasa es el estrés oxidativo en las mitocondrias neuronales, provocado por la acumulación intracelular de especies de oxígeno reactivo, al que me he referido anteriormente.
De nuevo, es el laboratorio de Busciglio el que analiza la cuestión de forma directa y aporta datos de hondo calado que voy a resumir (Busciglio et al., 2002). En su modelo de cultivos de células obtenidas de cerebros fetales con SD, antes descrito, obtienen los siguientes resultados:
A) Corteza fetal SD, cultivos de astrocitos. En los astrocitos SD:
- El nivel de APP está aumentado en relación con los no-SD.
- El nivel de C83 (que depende la actividad de la α-secretasa) está disminuido, mientras que el de C99 (que depende de la ß-secretasa) está aumentado. Es decir, se aprecia ya la desviación hacia el procesamiento patológico de la APP, con aumento selectivo de la actividad de la ß-secretasa y disminución de la α-secretasa.
- Los astrocitos SD muestran disminución de la secreción de APP y Aß, con acumulación intracelular de Aß en forma de agregados insolubles dentro de compartimientos subcelulares asociados con el tráfico y procesamiento de APP.
B) Corteza fetal Síndrome de Down, cultivos de neuronas. Antes del 5º día de cultivo, aumenta la presencia de APP, hay acumulación intracelular de Aß, con disminución de la secreción de Aß y APP.
C) La depleción energética provocada por un veneno mitocondrial en los astrocitos no-SD incrementó la presencia intracelular de APP y C99, mientras que redujo la concentración de C83 y APPs solubles. Estos cambios se parecen a lo observado en los astrocitos SD. Igualmente, se observaron agregados de Aß42 a todo lo largo y ancho del citoplasma.
D) Se comprueba que en los astrocitos SD existe una alteración del metabolismo energético mitocondrial. En efecto:
- El estudio del potencial transmembrana en la mitocondria de los astrocitos SD muestra que hay una disminución de este potencial.
- El estudio de la actividad redox en la mitocondria de los astrocitos Síndrome de Down demuestra una disminución.
E) En cerebros humanos:
- En el cerebro SD, tanto en neuronas como en astrocitos, existe acumulación intracelular de Aß42; esto puede preceder a la formación de placas amiloides y ovillos neurofibrilares.
- En cambio, hay una reducción de APP soluble.
- En neuronas corticales Síndrome de Down en cultivo, la incorporación de APP impidió la neurodegeneración. Esto no ocurrió en neuronas normales, lo que puede sugerir que poseen un valor neuroprotector.
Para conocer mejor el papel patogénico que puede jugar el estrés oxidativo cuando se combina con un incremento de formación de APP, tienen particular interés los resultados obtenidos en ratones doble-transgénicos que sobreexpresan dos genes: el SOD1 y el APP (Harris-Cerruti et al., 2004). Se sabe que la sobreexpresión de sólo la enzima SOD1 en ratones transgénicos produce aumento de los niveles de H2O2 y de radicales hidroxilo, con peroxidación de lípidos y lesión neuronal, dificultades en la memoria espacial y en los fenómenos de LTP. En el ratón doble-transgénico APP-SOD1 se apreció una fuerte acumulación de varias formas de APP sin que se apreciara aumento de Aß42. Por otra parte, los ratones mostraron, en función creciente de la edad, intensas alteraciones en el aprendizaje, en la memoria operacional y a largo plazo, así como en el fenómeno de la LTP. Se observó igualmente acumulación de lipofuscina y diversas anomalías en las mitocondrias. No se apreció, en cambio, ni gliosis reactiva ni pérdida neuronal. Estos resultados indican que no basta la sobreexpresión de APP y SOD1 (hechos que se aprecian en el SD) para que surja toda la neuropatología propia de la EA.
15.2.2.2.3 La influencia de la apolipoproteína E y su alelo ε4
Como ya se explicó, el gen de la apoE muestra varios alelos. La presencia del alelo ε4 es un importante factor de riesgo para el desarrollo de la enfermedad de Alzheimer (Polvikoski et al., 1995), y se ha observado que contribuye a la instauración de la demencia tipo Alzheimer en las personas con SD (Schupf et al., 1996). El alelo ε4 afecta al procesamiento de Aß: su presencia parece incrementar la deposición de las proteínas Aß40 y Aß42 en los cerebros (McNamara et al., 1998). De hecho, Schupf et al., (2001) apreciaron que los niveles plasmáticos de Aß42 estaban más aumentados en los adultos con Síndrome de Down que mostraban signos de demencia que en los que no la mostraban, y que la presencia del alelo ε4 mostraba asociación con la elevación de los niveles Aß42 y no con los de Aß40.
Se desconoce todavía el mecanismo por el que el alelo ε4 de la apoE facilita la mayor presencia y deposición del amiloide Aß42; si se trata de una mayor velocidad en la formación de fibrillas del amiloide, o de una menor velocidad en la limpieza y desaparición de dichas fibrillas. Pero lo más interesante, desde la perspectiva que estamos contemplando aquí, es la demostración en cultivos de células musculares lisas de que la presencia concurrente de estrés oxidativo favorece la acumulación de apoE4 y la formación de depósitos apoE-Aß; estos depósitos, a su vez, favorecen la aparición de peroxidación de lípidos (Mazur-Kolecka et al., 2003).
15.2.2.2.4 El papel de la reacción inflamatoria
Es frecuente observar una reacción inflamatoria alrededor de las placas neuríticas y de los depósitos de Aß. Esta reacción puede ser mayor en el SD y contribuir en mayor proporción a la progresión acelerada a partir de los 40 años. La acumulación de microglia y su consiguiente acción de fagocitosis puede acelerar la oxidación de Aß y facilitar una mayor formación de placas neuríticas. Además de la activación microglial hay un mayor grado de astrocitosis. En conjunto, pues, las neuronas, la microglia y los astrocitos pueden sintetizar y liberar factores inflamatorios (citoquinas, factores de crecimiento) que faciliten aún más el reclutamiento de monocitos, con la consiguiente formación de placas neuríticas y de la actividad reactividad que las acompaña (Lott y Head, 2005).
15.2.2.3 Una primera conclusión
De los datos expuestos podemos concluir lo siguiente. Es posible que el trastorno en el metabolismo energético de las células SD, consecuencia del estrés oxidativo al que se ven sometidas, sea un factor que contribuya a promover la actividad de la ß-secretasa, y de ese modo se facilite la ruptura de la molécula APP en el lugar inadecuado; además, altera el tráfico de Aß42 de modo que se acumula intracelularmente y forma agregados insolubles que ejercen su acción neurotóxica.
Es posible que esta acumulación intracelular, tan precozmente iniciada, tenga que ver con otro fenómeno que se ha descrito recientemente: la presencia intraneuronal de grandes endosomas. Estas estructuras intracelulares son ricas en proteasas (incluida la ß-secretasa), e intervienen en los procesos de intercambio, reciclaje y modulación catabólica de diversas macromoléculas. Se ha comprobado la presencia de endosomas anómalos en las primeras etapas de la enfermedad de Alzheimer; su actividad contribuye al procesamiento anormal de la APP por parte de la ß-secretasa, y se ha asociado de hecho la presencia de estos endosomas con la producción excesiva de Aß. Pues bien, también se ha demostrado que el mismo tipo de endosomas anómalos se encuentra muy precozmente en las neuronas del SD (Cataldo et al., 2000), incluso antes del nacimiento, en estructuras cerebrales como son el hipocampo, la corteza y los ganglios de la base.
No se descarta la posibilidad de que la propia sobreexpresión de APP en el SD sea uno de los factores que alteren la función mitocondrial; o que el mismo Aß42 por su acción tóxica sea la que altere la función mitocondrial. Es también posible que el exceso de actividad necesaria para aclarar o limpiar los agregados de proteínas suponga un fuerte coste metabólico para la célula debido a la cantidad de energía que requiere, y eso facilite la alteración de la actividad mitocondrial.
Por último, la existencia de un alelo ε4 constituye un factor adicional que, aunque no sea determinante, contribuye a la iniciación de la amiloidogénesis patológica. El propio estrés oxidativo puede facilitar la acción patogénica de la apolipoproteína E4.
15.2.2.4 Consecuencias
A partir de todo lo expuesto hasta aquí, parece razonable avanzar algunas hipótesis. Hay una actividad de fondo caracterizada por la presencia de estrés oxidativo en las células de los organismos con SD. Su intensidad puede ser muy variable dependiendo del grado en que se sobreexprese el gen de la SOD1, así como de la presencia de otros factores que pueden contribuir a su mantenimiento, como puede ser el exceso de moléculas APP. Esa acción es permanente, y su influencia se expresa sobre las células de distintos tejidos a lo largo de la vida, a las que somete a un desgaste constante que puede explicar la precocidad con que aparece el envejecimiento en el SD.
Al mismo tiempo, la lesión de las estructuras mitocondriales, inherente al estrés oxidativo, parece ser capaz de desencadenar la activación de la vía de la ß-secretasa, junto con otros factores coadyuvantes. Y sobre el fondo de una sobreproducción de APP debida a la sobreexpresión del gen APP presente en el cromosoma 21, se dan las condiciones para ir produciendo a lo largo de los años el exceso de Aß42 neurotóxico. La intensidad con que esta proteína se produce, la velocidad con que se genera, y la distribución por los núcleos y áreas cerebrales, van a ser los factores determinantes de la aparición de los signos y síntomas propios de la demencia Alzheimer, de la precocidad con que aparezca, de la intensidad de sus manifestaciones, y de la rapidez de su evolución.
De lo expuesto se deduce el papel crítico que la alteración de los mecanismos oxidativos desempeña en ambos procesos: la precocidad del envejecimiento y la evolución hacia la enfermedad de Alzheimer. Por el mismo motivo, una intervención dirigida a contrarrestar y controlar los procesos oxidativos anormales podría tener unas notables y beneficiosas consecuencias.
De nuevo, el laboratorio de Busciglio nos ofrece una posible pista (Pelsman et al., 2003). Su grupo de investigación ha examinado la actividad de un compuesto que es análogo dipéptido del piracetam, el GVS-111 (DVD-111), para contrarrestar la acción neurotóxica del peróxido de hidrógeno sobre las neuronas de la corteza cerebral fetal mantenidas en cultivo. En las neuronas normales, el producto fue capaz reducir la mortalidad y toxicidad ocasionadas por el superóxido. En las neuronas de fetos con SD, redujo y retrasó la aparición habitual de cambios degenerativos, y redujo la acumulación de radicales libres intracelulares. En nuestro laboratorio, estamos analizando la posible influencia de este producto en el ratón Ts65Dn, un modelo de Síndrome de Down.
Finalmente, vistos los evidentes puntos de contacto entre la enfermedad de Alzheimer y el envejecimiento en el Síndrome de Down, cuanto terapéuticamente sea útil para prevenir o aliviar la primera lo será también para prevenir o aliviar el peculiar proceso de envejecimiento en el Síndrome de Down, como se ha podido comprobar en el caso de los inhibidores de la acetilcolinesterasa (Prasher et al., 2002).
Referencias
- Bertram L., Tanzi R. E. Alzheimer’s disease: one disorder, too many genes? Hum. Mol. Genet. 2004;13:135-141.
- Braak H., Braak R. Patterns of cortical lesions in Alzheimer’s disease. En: Alzheimer’s disease: biology, diagnosis and therapeutics. Iqbal K., Winblad B., Nishimura T., Takeda M., Wisniewski H. M. (editores). John Wiley & Sons Ltd. London 1997;227-237.
- Busciglio J., Yankner B. A. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 1995;378:776-779.
- Busciglio J., Pelsman A., Wong C., Pigino G., Yuan M., Nori H., Yankner B. A. Altered metabolism of the amyloid ß precursor protein is associated with mitochondrial dysfunction in Down’s syndrome, Neuron 2002;33:677-688.
- Capone G., Kim P., Jovanovich S., Payne L., Freund L., Welch K., Miller E., Trush M. Evidence for increased mitochondrial superoxide production in Down syndrome. Life Sci. 2002;70:2885-2895.
- Cataldo A. M., Peterhoff C. M., Troncoso J. C., Gomez-Isla T., Hyman B. T., Nixon R. A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000;157:277-286.
- Cataldo A. M., Petanceska S., Peterhoff C. M., Terio N. B., Epstein C. J., Villar A., Carlson E. J., Staufenbiel M., Nixon R. A. App. gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of Down syndrome. J. Neurosci. 2003;23:6788-6792.
- Caughey B., Lansbury P. T. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsable protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003;26:267-298.
- Chicoine B., McGuire D. Longevity of a woman with Down syndrome: a case study. Ment. Retard 1997;35:477-479.
- Engidawork E., Lubec G. Molecular changes in fetal Down syndrome brain. J Neurochemistry 2003;84:895-904.
- Flórez J. Síndrome de Down y envejecimiento. En: http://www.down21.org/salud/salud/Envejecimiento_y_SD.htm. 2002.
- Flórez J. Síndrome de Down: presente y futuro. Rev. Síndrome Down 2003a;20:16-22.
- Flórez J. Farmacología de las demencias y de las conductas anormales. En: Farmacología Humana. Flórez J., Armijo J. A., Mediavilla A. (editores). Masson. Barcelona. 2003b; 623-634.
- Flórez J., Ruiz E. Síndrome de Down. En: Síndromes específicos e individualidad de los apoyos: un enfoque interdisciplinar. Del Barrio J. A., Borragán A. (editores). Universidad de Cantabria, Departamento de Educación. Santander. 2003;41-57.
- German D. C., Eisch A. J. Mouse models of Alzheimer’s disease: insiight into treatment. Rev. Neurosci. 2004;15:353-369.
- Harris-Cerruti C., Kamsler A., Kaplan B., Lamb B., Segal M., Groner Y. Functional and morphological alterations in compound transgenic mice overexpressing Cu/Zn superoxide dismutase and amyloid precursor protein. Eur. J. Neurosci. 2004;19:1174-1190.
- Kim S. H., Vikolinsky R., Cairns N., Lubec G. Decreased levels of complex III core protein 1 and complex V beta chain in brain from patients with Alzheimers’s disease and Down syndrome. Cell. Mol. Life Sci. 2000;57:1810-1816.
- Klein W. L., Krafft G. A., Finch C. E. Targetting small Aß oligomers; the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219-224.
- Lemere C. A., Blusztajn J. K., Yamaguchi H., Visniewski T., Saido T. C., Selkow D. J. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 1996;3:16-32.
- Lott I. T., Head E. Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol. Aging 2005;26:383-389.
- Lue L. F. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimers’s disease. Am. J. Pathol. 1999;155:853-862.
- Mazur-Kolecka B., Kowal D., Sukontasup T., Dickson D., Frackowiak J. The effect of oxidative stress on accumulation of apolipoprotein E3 and E4 in a cell culture model of ß-amyloid angiopathy (CAA). Brain Res. 2003;983:48-57.
- McNamara M. J., Gomez-Isla T., Hyman B. T. Apolipoprotein E genotype and deposits of Abeta40 and Abeta42 in Alzheimer disease. Arch. Neurol. 1998;55:1001-1004.
- Mehta P. D., Dalton A. J., Mehta S. P., Kim K. S., Sersen E. A., Wisniewski H. M. Increased plasma amyloid beta protein 1-42 levels on Down syndrome. Neurosci. Let. 1998;241:13-16.
- Mori C., Spooner E. T., Wisniewski K. E., Wisniewski T. M., Yamaguchi H., Saido T. C., Tolan D. R., Selkoe D. J., Lemere C. A. Intraneuronal Aß42 accumulation in Down syndrome brain. Amyloid: J. Protein Folding Disord. 2002;9:88-102.
- Pascual J. Enfermedades degenerativas del sistema nervioso. En: Medicina Interna. Rodés J., Guardia J. (editores). Masson. Barcelona. 2004;1998-2017.
- Pelsman A., Hoyo-Vadillo C., Gudasheva T. A., Seredenin S. B., Ostrovskaya RU, Busciglio J. GVS-111 prevents oxidative damage and apoptosis in normal and Down’s syndrome human cortical neurons. Int. J. Devl. Neuroscience. 2003;21:117-124.
- Polvikoski T., Sulkava R., Haltia M., Kainulainen K., Vuorio A., et al. Apolipoprotein E., dementia and cortical deposition of beta-amyloid protein. N. Eng. J. Med. 1995;333: 1242-1247.
- Prasher V. P., Huxley A., Haque M. S. and the Down syndrome Ageing Study Group. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Down syndrome and Alzheimer’s disease – Pilot Study. Int. J. Geriat. Psychiatry 2002;17: 270-278.
- Rang H. P., Dale M. M., Ritter J. M., Moore P. K. Farmacología, 5ª edic. Elsevier España. Barcelona. 2004;495.
- Schupf N., Kapell D., Lee J. H., Zigman W., Canto B., Tycko B., Mayeux R. Onset of dementia is associated with apolipoprotein E epsilon 4 in Down’s syndrome. Ann. Neurol. 1996;40:799-801.
- Schupf N., Patel B., Silverman W., Zigman W. B., Zhong N., Tycko B., Mehta P. D., Mayeux R. Elevated plasma amyloid ß-peptide 1-42 and onset of dementia on adults with Down syndrome. Neurosci. Let. 2001;301:199-203.
- Selkoe D., Kopan R. Notch and presenilins: regulated intramembrane proteolysis links development and degeneration. Annu. Rev. Neurosci. 2003;26:565-597.
- Teller J. K., Russo C., DeBusk L. M., Angelini G., Zaccheo D., Dagna-Bricarelli F., Scartezzini P., Bertolini S., Mann D. M., Yabaton M., Gambetti P. Presence of soluble amyloid beta peptide precedes amyloid plaque formation in Down’s syndrome. Nat. Med. 1996;2:93-95.
- Tokuda Y., Fukushima T., Ikeda S-I., Sekijima Y., Shoji S., Yanagisawa N., Tamaoka A. Plasma levels of amyloid ß protein Aß 1-40 and Aß 1-42(43) are elevated in Down’s syndrome. Ann. Neurol. 1997;41:271-273.